Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Факторы динамики генетического состава популяции. Дрейф генов, мутационный процесс, миграции, избирательное спаривание особей, естественный отбор.Содержание книги
Поиск на нашем сайте
М утагены окружающей среды: Биологические мутагены: специфические последовательности ДНК—транспозоны; некоторые вирусы (вирускори,краснухи,гриппа);продукты обмена веществ(продуктыокисления липидов); Физическими мутагенами называются любые физические воздействия на живые организмы, которые оказывают либо прямое влияние на ДНК или вирусную РНК, либо опосредованное влияние через системы репликации, репарации, рекомбинации- это разные виды излучений: ионизирующее излучение, радиоактивный распад, ультрафиолетовое излучение. Химическим мутагенам относятся многие химические соединения самого разнообразного строения. Наибольшую мутагенную активность проявляют различные алкилирующие соединения, а также нитрозосоединения, некоторые антибиотики, обладающие противоопухолевой активностью. 3. Геномика - наука о геномах. Структурная организация генома прокариот и эукариот. Классификация повторяющихся элементов генома. Международный проект «Геном человека». Геномика — раздел молекулярной генетики, посвящённый изучению генома и генов живых организмов. Прокариоты характеризуются отсутствием клеточного ядра, отделенного мембраной от цитоплазмы. Генетический аппарат представлен одной или несколькими кольцевыми молекулами ДНК. Однако сейчас описаны виды с линейными молекулами ДНК, а также виды у которых кольцевая молекула сочетается с линейной. Для клеток эукариот характерно наличие оформленного ядра. Информационной макромолекулой их генома является ДНК, которая неравномерно распределена по нескольким хромосомам в виде комплексов с многочисленными белками. Однако генетическую информацию в клетках содержат не только хромосомы ядра. Жизненно важная генетическая информация заключена и во внехромосомных молекулах ДНК. У эукариот — это ДНК хлоропластов, митохондрий и других пластид. Под геномом эукариотического организма в настоящее время понимают суммарную ДНК гаплоидного набора хромосом и каждого из внехромосомных генетических элементов, содержащуюся в отдельной клетке зародышевой линии многоклеточного организма. Под повторяющимися последовательностями ДНК (повторами) понимается широкий спектр как умеренно повторяющихся, так и часто повторяющихся (высокоповторяющихся) последовательностей ДНК. По крайней мере 20—30% генома человека представлено повторами.
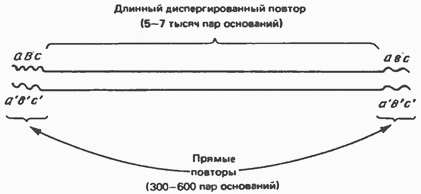
Высокоповторяющиеся последовательности состоят из участков длиной в 5—500 пар оснований, повторяющихся много раз и расположенных один за другим (тандемно). Эти последовательности обычно образуют кластеры и присутствуют в количестве от 1 до 10 миллионов копий на гаплоидный геном. Высокоповторяющиеся последовательности транскрипционно-неактивны и, вероятно, участвуют в структурировании хроматина. Умеренно повторяющиеся последовательности, присутствующие в количестве менее чем 106 копий на гаплоидный геном, не образуют кластеров, а чередуются с неповторяющимися (уникальными) последовательностями. Они могут быть как короткими, так и весьма протяженными. Длинные диспергированные повторы состоят из 5000—7000 пар оснований и представлены в количестве 1000—100000 копий на гаплоидный геном. Они фланкированы с обоих концов прямыми повторами длиной в 300— 600 пар оснований (рис. 38.8). Во многих случаях длинные повторы транскрибируются РНК-полимеразой II в виде молекул мРНК, содержащих такие же кэпированные 5-концы, как и мРНК.
Схема длинного диспергированного повтора. Отмечено расположение на концах повтора коротких прямых повторов (abc) и соответствующих комплементарных участков (аbс).Короткие диспергированные повторы представляют семейства родственных, но отличных друг отдруга фрагментов длиной от единиц (нескольких пар) до нескольких сотен пар нуклеотидов. Проект Человеческий Геном — международный научно-исследовательский проект, главной целью которого было определить последовательность нуклеотидов, которые составляют ДНК, и идентифицировать 20—25 тыс. генов в человеческом геноме[1]. Этот проект называют крупнейшим международным сотрудничеством, когда-либо проводившимся в биологии[2]; он стал основой для международного проекта GenomeProject-write. целью проекта по расшифровке генома человека является понимание строения генома человеческого вида, проект также фокусировался и на нескольких других организмах, среди которых бактерии, в частности, насекомые, такие как мушка дрозофила, и млекопитающие, например, мышь. 4. Популяционно-статистический метод. Генетическая структура популяции. Частоты генов и генотипов. Закон Харди-Вайнберга. Г енетический груз.
Метод находит широкое применение в клинической генетике, т.к. внутрисемейный анализ заболеваемости не отделим от изучения наследственной патологии как в станах с большим населением, так и в относительно изолированных популяционных группах. Сущность метода заключается в изучении (с помощью методов вариационной статистики) частот генов и генотипов в различных популяционных группах, что дает необходимую информацию о частоте гетерозиготности и степени полиморфизма у человека. В частности, в гетерозиготном состоянии в популяциях находится значительное количество рецессивных аллелей, что обуславливает развитие различных наследственных заболеваний, частота которых зависит от концентрации рецессивного гена в популяции и значительно повышается при заключении близкородственных браков. Мутации могут передаваться потомству во многих поколениях, что приводит к генетической гетерогенности, лежащей в основе полиморфизма популяций. Согласно закону Харди-Вайнберга (1980) - в популяции сохраняется постоянное соотношение чатоты генотипов из поколения в поколение, если никакие факторы не нарушают это равновесие. Формула Харди-Вайнберга:Р - частота, с которой встречается доминантный ген "А"; g- частота, с которой встречается рецессивный аллель "а". Сумма Р+g всегда равна 1 Соотношение генотипов АА, Аа, аа выражается формулой: (Р+g)2 =Р2+2Hg+g2. Статистический анализ распространенности отдельных генов и контролируемых ими признаков в популяционных группах позволяет определить адаптивную ценность конкретныхгенотипов. Среди людей невозможно найти генетически одинаковых лиц (за исключением монозиготных близнецов, для которых предполагается 100% общих генов), хотя общность генов хорошо прослеживается у близких и дальних родственников.
Принцип. Получение рекомбинантных ДНК. Для получения таких молекул первоначально выделяют ДНК из 2 разных источников. Каждую из них в отдельности фрагментируют, используя одну и ту же рестриктазу, расщепляющую ДНК с образованием "липких" концов. После процедуры нагревания и медленного охлаждения наряду с исходными молекулами могут образовываться рекомбинантные молекулы, состоящие из фрагментов связанных между собой "липкими" концами. Клонирование ДНК. Для получения значительных количеств интересующего нас материала проводят клонирование ДНК, предполагающее встраивание нужного нам фрагмента ДНК в векторную молекулу ДНК (или вектор). Вектор обеспечивает проникновение этой рекомбинантной, ДНК в бактериальные клетки. В качестве векторов используют плазмиды, фаги, ретро- и аденовирусы. Особенно часто в качестве вектора служит плазмидная ДНК. Принцип. Картирование генома. Весь геном сначала случайно разбивается на более мелкие последовательности ДНК - приблизительно от 100 000 до 300 000 пар оснований. Эти участки затем клонируют в бактериальном векторе. Повторяя этот цикл несколько раз, исследователи получают серию перекрывающихся ДНК. Затем эти пропускают через гель-электрофорез, чтобы определить их индивидуальные последовательности.
Секвенирование ДНК.
Анализ результатов. Несколько перекрывающихся последовательностей ДНК используется для определения порядка фрагментов в каждом.
В этой процедуре используется несколько различных компьютерных программ, которые могут анализировать последовательности ДНК. 12. Рестрикционный анализ. Сущность метода заключается в обработке ДНК рестрикционными ферментами (специфическими эндонуклеазами), разрезающими молекулу ДНК по определенным последовательностям нуклеотидов. После этого анализируют полученные фрагменты, специфические для каждого вида или варианта микроорганизма, а также на наличие мутации. Заболевания (МФЗ). Основу патогенеза МФЗ составляет общебиологическое явление изменчивости, сбалансированный наследственный полиморфизм, т.е. наличие в популяции 2-х и более фенотипов, один из которых считается нормальным, а другие (более редкие) - вариантными.
16. Близнецовый метод, цели, этапы, расчет коэффициента Хольцингера близнецового метода 1. Подбор близнецовых пар: монозиготных (MZ) и дизиготных (DZ). 2. Расчет конкордантности для монозигот и дизигот.
С -число конкордантных пар; D -число дискордантных пар. 3 Расчет коэффициента наследуемости (Хольцингера):
Генные болезни. Общие понятия. Классификация. Примеры. Генные болезни - это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена Виды генных мутаций: · замена пар нуклеотидов (миссенс- мутации и нонсенс- мутации) · выпадения (делеция) одной или нескольких пар нуклеотидов · вставка нуклеотида (инсерция) · экспансия тринуклеотидных повторов · нарушение сплайсинга Результатом миссенс- мутации являются: · серповидно-клеточная анемия (замена глутаминовой кислоты в молекуле HbA на валин в 6 положение в молекулах HbS) · различия по инсулину: треонин в 30 положение b-цепи- инсулин человека; аланин- инсулин свиньи; серин- инсулин кролика; a-цепь у всех одинакова · a-b- талассемия · иммуноглобулинjпатия · муковисцидоз Dставки (инсерции) вызывают аналогичные изменения рамки считывания только в другую сторону пр.миодистрофияДюшенна и Беккера, муковисцидоз Экспансия тринуклеотидных повторов -в норме число одинаковых триплетов в нити ДНК постоянно, при экспансии тринуклеотидных повторов их чего может увеличиваться в несколько раз Пр. · хорея Гентингтона (в 4 хромосоме число триплетов ЦАГ увеличивается в 3 раза) · миотическая дистрофия (в 19 хромосоме число повторов ЦТГ увеличивается в 400 раза и более) · синдром fra-x- умственная отсталость с ломкой x-хромосомой (триплет ЦГГ повторяется от 200 до 2000 раз (в норме 6-51 раз))
· спинно- церебральная атаксия в 6 хромосоме триплет ЦАГ повторяются до 80 раз (в норме 25-36) Мутация в одной из клеток на ранних стадиях дробления приводит к возникновению мозаичныхформ (обнаружены при миопатии Дюшенна) Если мутации в клетках половых органов- мозаицизм гонад Частота мозаицизм у родителей составляет 5-15% случаев в доминантных и Х- сцепленных рецессивных болезней: · несовершенный остеогенез · синдром Элерса-Данло · гемофилия 22. Клинико-генеалогический метод. Типы наследования. Клинические примеры. Клинико-генеалогический метод позволяет: · выявить наследственный характер заболевания · установить тип наследования болезни · установить зиготность членов членов родословной · определите вероятность появления признаков в потомстве · дать обоснованный прогноз для потомства в семье, где есть носитель патологического гена · изучить гетерозиготность наследственных болезней Показания: · наличие у больного и его родственников моногенного заболевания · заболевания с наследственной предрасположенностью · семейный характер заболевания · непереносимость некоторых пищевых продуктах · извращенная реакция на действие лекарств · аллергические заболевания в семье · кровное родство родителей больного ребенка · отягощенный акушерский анамнез · врождённые пороки развития Клинико-генеалогический метод лежит в основе медико-генетического консультирования и включает 3 этапа: 1 этап- сбор клинического анамнеза и клиническое обследование 2 этап- составление родословной 3 этап- генетический анализ родословной Анамнез настоящего заболевания (начало, характер, течения) · Рецидивирующая, хроническая, длительно не поддающаяся лечению течения, особенно в детском возрасте (хроническая пневмония при муковисцидозе, расстройство кишечника при целиакии, дисахаридазная недостаточность) · стойки изменения в моче (протеинурия, гематурия)- синдром Альпорта или семейная гематурия · стойкая гипотрофия (муковисцидоз, целиакия, дисахаридазная недостаточность) · прогрессирующее, не поддающиеся лечению судорожные приступы · наличие у больного редких, специфических симптомов или их сочетаний · вовлечение в патологический процесс многих органов и систем. Ообенно распространённый характер поражения наблюдается при хромосомной патологии Анамнез жизни: · течение беременности и родов, периода новорождённости, вскармливание, физическое и нервно-психическое развитие, перенесённые заболевания, данные акушерского анамнеза · сведения о сибсах · сведения о родителей · национальность (болезнь Гоше, болезнь Ниманна-Пика у евреев, некоторые формы наследственных аниме у негров) · места жительства семьи (влияние эндемического фактора) · места жительства предков (выявление кровно родственных браков) · профессия (профессиональные вредности, приём лекарственных препаратов во время беременности- возможность тератогенного действия на плод)
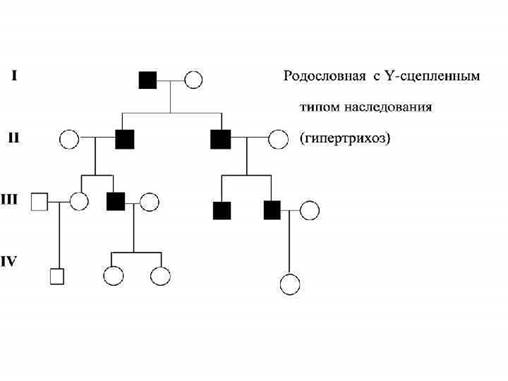
Объективное обследование пробанда: · детальный осмотр и его и родственников · антропометрия · описание фенотипических проявлений заболевания Синдром Элерса-Данло, нейрофиброматоз Патология мышечной ткани: · характерно во многих наследственных синдромов · аплазии ряда мышцы верхних конечностей (при синдроме Эдвардса) Патология костной системы: · деформация грудной клетки, черепа и позвоночника, слегка согнутые конечности, широкие · хрупкие кости, множественное спонтанные переломы- несовершенный остеогенез · длинные, тонкие паучьи пальцы, изменённое грудная клетка- патогномоничный признак болезни Марфана Антропометрические исследования: · рост · массы тела · телосложения · длина конечностей · окружность груди и головы · соотношения сагиттального энтерального размеров черепом Высокий рост, длины конечности, арахнодактилия указывают на синдром марфана Укорочении конечностей по сравнению с туловищем, запавшая переносица- ахондроплазия. Микроцефалия- симптомы их наследственных болезней 23. Особенности аутосомно-доминантного типа наследования. Составить родословную. Аутосомно-доминантный тип наследования характеризуется: · патологический признак встречается в каждом поколении и проявляется у гетерозигот; соотношение здоровых и больных чипсов приближается 1: 1 · чаще неполная пенетрантность признака (пр. пенетрантность для ретинобластомы примерно 90%) · различная выраженность клинических проявлений не только между разными семьями, но и внутри каждой семьи (пр.нейрофиброматоз у одних членов семьи- генерализованная форма, а у других- отдельные кожные проявления; синдром марфана) · клинические проявления могут развиваться спустя несколько лет после рождения, а сроки проявления у разных членов семьи разные (пр. хорея Гентингтона, миопическая дистрофия, которое может иметь начала от пренатального периода до 50 60 лет) Близорукость Аутосомно-рецессивный тип наследования характеризуется: · риск рождения больного ребенка 25%, если родители здоровы и гетерозиготное · если родители гомозиготны, то все сибсы должны быть больными (пр. альбинизм, фенилкетонурия) · браке гетерозигот с гомозиготами дают менделеевское расщепление больных и здоровых 1: 1 Это главным образом кровнородственные браки По аутосомно рецессивному признаку наследуется в основном энзимопатии, многие из них с известными биохимическим дефектом Особености Y сцепленного типа наследования. Составить родословную. Примеры заболеваний. 1. Гены расположены в Y хромосоме 2. Болезнь передается только от отца 3. Заболевание наследуется только мальчиками 4. Признак наблюдается в каждом поколение по отцовской линии 5. Примеры: гипертрихоз ушной раковины, перепонка между пальцами.
Особенности Х сцепленного доминантного типа наследования. Родословная. Примеры. 1. если болен отец, то все его дочери- больные а все сыновья, здоровые, 2. больными будут дети только в том случае, если болен один из родителей 3. у здоровых родителеи все дети будут здоровы 4. заболевание прослеживается в каждом поколении 5. если мать больна, то вероятность рождения больного ребенка равна 50 % независимо от пола; 6. болеют как мужчины,так и женщины, но в целом больных женщин в семье в 2 раза больше, чем больных мужчин. 7. Например, Витамин - Д-резистентный рахит
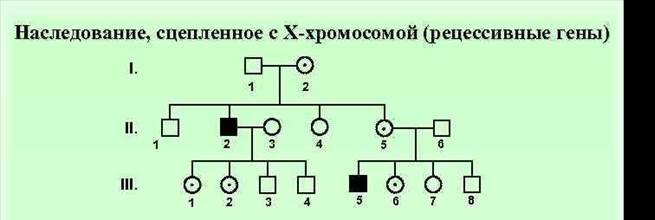
Особенности Х сцепленного рецессивного типа наследования. Родословная. Примеры. 1. преимущественно поражаются мужчины 2. признак проявляется фенотипически не в каждом поколении, поэтому от брака больного мужчины и здоровой женщины все дети здоровы 3. заболевание наблюдается у мужчин - родственников пробанда по материнской линии. 4. сын никогда не наследует болезни отца; 5. при браке между здоровым мужчиной и гетерозиготной женщиной вероятность рождения больного ребенка составляет 25 % для мальчиков и 0 % для девочек 6. Так наследуется гемофилия, дальтонизм,мужское бесплодие
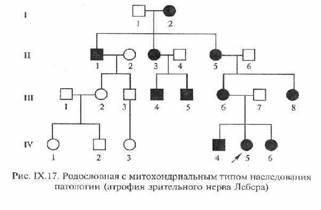
29. Передача генетической информации через цитоплазму получила название цитоплазматической (внеядерной, нехромосомной) наследственности. Поскольку наследственная информация передается по материнской линии через цитоплазму яйцеклетки, ее называют также материнской наследственностью. Непосредственное влияние материнского организма на развитие зародыша часто приводит к большему сходству потомства с матерью, поскольку условия эмбрионального развития организма полностью зависят от матери.

30. Наследственные болезни обмена – группа моногенных наследственных заболеваний, обусловленных мутациями генов, кодирующих ферменты, транспортные или сигнальные белки, что приводит к нарушению метаболизма клетки. Этиология Аутосомно - рецессивное наследственное заболевание, связанное с мутацией гена, расположенного на длинном плече хр. 7 в области q31– q32. Генетика Структура гена муковисцидоза) полностью расшифрована. Длина гена около 250 000 пар оснований. Несмотря на большой размер гена, число выявленных мутаций ограничено, что свидетельствует о малом мутационном вкладе в развитие заболевания. До 70% мутаций у больныхr(219700, 7q31. 2, CFTR, CF, муковисцидозом в некоторых европейских популяциях составляет делеция 3 пар оснований, приводящая к потере фенилаланина в 508 положении предполагаемого продукта гена. Вклад других мутаций различен в разных популяциях и приводит к клиническому разнообразию симптоматики. Патогенез Генетический дефект каналов для ионов хлора. Нарушение транспорта хлора ведёт к нарушению транспорта воды и дегидратации секрета • Вязкий секрет закупоривает выводные протоки желёз • Развивается воспалительный процесс с присоединением вторичной инфекции. Нарушение транспорта Na и CI через клеточные мембраны. Это приводит к избыточному выведению хлоридов, что проявляется гиперсекрецией густой слизи в клетках эндокринной части поджелудочной железы, эпителии бронхов и ж.к.т. Проявляется в 4 формах: 1.Меконеальный илеус новорожденных, 2. Кишечная форма; 3.Бронхолегочная и 4.Смешанная форда.
Муковисцидоз - (от латинского mucus- слизь, viscidus- вязкий) достаточно распространенное наследственное заболевание, при котором поражаются все органы, которые выделяют секреты. Это бронхолегочная система, поджелудочная железа, печень, потовые железы слюнные железы, железы кишечника, половые железы. Из-за дефекта (мутации) гена секреты во всех органах вязкие густые, поэтому их выделение затруднено. В легких из-за вязкого, часто гнойного секрета, трудноотделяемого и скапливающегося в бронхах, довольно быстро (иногда уже в первые месяцы жизни) развиваются воспалительные процессы - повторные бронхиты и/или пневмонии с постепенным формированием хронического бронхолегочного процесса. У детей отмечаются постоянный раздражающий (иногда коклюшеподобный) кашель, одышка. Из-за недостатка ферментов поджелудочной железы у больных муковисцидозом плохо переваривается пища, поэтому такие дети, несмотря на повышенный аппетит, отстают в весе, у них обильный, жирный зловонный стул. Из- за застоя желчи у некоторых детей развивается цирроз печени, могут сформироваться камни в желчном пузыре. Каждый 20-й житель планеты является носителем дефектного гена. Муковисцидоз возникает у ребенка в том случае, если он от обоих родителей (и от мамы, и от папы) получил по гену с мутацией Никакие болезни родителей, их курение или прием алкогольных напитков, стрессовые ситуации значения не имеют. Муковисцидоз одинаково часто встречается как у мальчиков, так и у девочек. Диагностика Основными методами диагностики муковисцидоза являются сбор анамнеза, клинические и лабораторные исследования. Поскольку заболевание является наследственным, важную роль при его обнаружении играет сбор информации о пациенте, наличии в его роду людей, являющихся носителями дефектного гена или болеющих муковисцидозом. Клиническое обследование позволяет обнаружить внешние признаки нарушений в работе определенных систем организма, свойственных данному заболеванию. С помощью лабораторных исследований специалисты смогут опровергнуть или подтвердить болезнь, а также оценить степень поражения организма. Фенилкетонурия
Фенилкетонурия ФКУ (синоним фенилпировиноградная олигофрения) - аутосомно-рецессивная болезнь аминокислотного обмена. В самостоятельную форму ФКУ выделил А.Фелинг в 1934году. (ФКУ) является наиболее распространенной аминоацидопатией. Частота ФКУ среди новорожденных по данным массового скрининга в различных странах составляет в среднем 1:10000. Была описана A. Foiling в 1934 г. Заболевание наследуется аутосомно-рецессивно и вызвано мутацией гена ФАГ. В патогенезе ФКУ имеют значение следующие обстоятельства: прямое токсическое действие на центральную нервную систему фенилаланина и его производных, нарушения в обмене белков, липо- и гликопротеидов, расстройства транспорта аминокислот, нарушение метаболизма гормонов и др., а также перинатальные факторы. Клиническая картина. Признаки фенилкетонурии обнаруживаются уже в первые недели и месяцы жизни Дети отстают в физическом и нервно-психическом развитии; отмечаются вялость, чрезмерная сонливость или повышенная раздражительность, плаксивость. По мере прогрессирования болезни могут наблюдаться эпилептиформные припадки - развернутые судорожные и бессудорожные типа кивков, поклонов, вздрагиваний, кратковременных отключений сознания. Гипертония отдельных групп мышц проявляется своеобразной "позой портного" (поджатые ноги и согнутые руки). Могут наблюдаться гиперкинезы, тремор рук. атаксия, иногда парезы по центральному типу. Дети нередко белокурые со светлой кожей и голубыми глазами, у них часто отмечаются дерматиты, экзема, повышенная потливость со специфическим (мышиным) запахом пота и мочи. Обнаруживается склонность к артериальной гипотензии. При отсутствии лечения развивается идиотия или имбецильность, глубокая психическая инвалидность. Лечение представляет собой диету со строгим ограничением поступления фенилаланина с пищей. Диета вводится с момента подтверждения диагноза классической ФКУ и длится минимум до полового созревания. Большинство иностранных врачей придерживается мнения о необходимости пожизненной диеты (long-lifediet). Диагностика Производится полуколичественным тестом или количественным определением фенилаланина в крови. При нелеченных случаях возможно выявление продуктов распада фенилаланина (фенилкетонов) в моче (не ранее 10—12 дня жизни ребёнка). Также возможно определение активности фермента фенилаланингидроксилазы в биоптате печени и поиск мутаций в генефенилаланингидроксилазы. Для диагностики 2 и 3 типа, связанных с мутацией в гене, отвечающем за синтез кофактора, необходимы дополнительные диагностические исследования. Общая характеристика митохондриальных патологий. Особенности генома митохондрий. Примеры заболеваний. Митохондриальные заболевания - большая группа патологических состояний, обусловленных нарушениями структуры и функции митохондрий по высвобождению энергии органических веществ и ее аккумуляции в виде макроэргических соединений. Отдельные формы митохондриальных заболеваний были давно известны врачам различных специальностей под названиями прогрессирующая офтальмоплегия, офтальмоплегия+, миопатия Изучение природы этих состояний началось в 60-е годы XX века. - общие закономерности митохондриальных заболеваний: 1. Материнский тип передачи 2. Феномен гетероплазмии. Клетки больного с митохондриальной патологией содержат мутантные и нормальные митохондрии, распределение которых происходит случайно при клеточном делении. 3. Зависимость тяжести клинических проявлении от характера мутационного повреждения митохондриального генома, содержания мутантной мтДНК в клетке, а также от энергетической потребности рамичных органов и тканей. 4. Высокая частота спорадических случаев. Скорость мутирования митохондриальной ДНК в 6-17 раз выше, чем ядерной ДН К, что обусловливает высокую частоту спорадических случаев митохондриальных заболеваний и определяет значительную роль мутаций митохондриальиого генома в возникновении хронических прогрессирующих и дегенеративных заболеваниях человека. Примеры Митох-х З-й: наследственная атрофия зрительных нервов (синдром Лебера), митохондриальная энцефаломиопатия, сахарный диабет с митохондриальным наследованием; Синдром Кирнса-Сейра и синдром Пирсона являются болезнями, обусловленными делециями митохондриальной ДНК.
Орфанные заболевания. Общие понятия. Примеры. Реестр орфанных заболеваний. Впервые термин «орфанные болезни» (редкие заболевания, «болезни-сироты») появился в 1983 г. в США при принятии закона об орфанных болезнях. В большинстве случаев редкие заболевания — это хронические болезни, требующие сложного и дорогостоящего лечения. Зачастую эти заболевания приводят к той или иной степени инвалидизации пациента, снижают качество жизни и ее продолжительность. Понятие орфанных заболеваний закреплено законодательно во многих странах, однако везде в него вкладывается несколько разный смысл. Дело в том, что одна и та же болезнь может быть крайне редкой в одной части мира, но при этом часто встречаться в других регионах. Например, болезнь Гоше для большинства этнических групп является редким заболеванием (1 случай на 50 тысяч новорожденных), однако значительно чаще встречается среди ашкеназских евреев.
Мукополисахаридоз — одна из групп тяжелых наследственных заболеваний обмена веществ. Ежегодно в России в среднем рождается до 10–17 детей с различными типами мукополисахаридоза.
Гемофилия — наследственное заболевание, которое характеризуется недостатком факторов свертывания крови, что приводит к замедленному ее свертыванию и удлинению времени кровотечения. В России зарегистрировано 8 тысяч больных гемофилией. Раньше все больные гемофилией уже к 15 годам становились инвалидами.
Болезнь Гоше — наследственное заболевание, развивающееся в результате недостатка фермента глюкоцереброзидазы, приводящего к избыточному накоплению липидов в костном мозге, печени, селезенке. Распространенность — 1 случай на 50 тыс. новорожденных. Болезнь имеет национальную доминанту, «отдавая предпочтение» ашкеназским евреям.
Болезнь Фабри — наследственная лизосомальная болезнь накопления (дефицит фермента α-галактозидазы), приводящая к инфаркту миокарда, хронической почечной недостаточности и, возможно, к летальному исходу. Мировая распространенность — 1 случай на 5–10 тыс. человек. Фенилкетонурия (дефицит гидроксилазы) — заболевание, связанное с нарушением обмена аминокислот и приводящее к поражению центральной нервной системы. Чаще встречается в северных европейских странах — частота 1:10 тыс., в Ирландии отмечается с частотой 1:4560, в России — 1:8–10 тыс., почти не встречается у представителей негроидной расы.
Синдром Элерса — Данлоса — группа заболеваний соединительной ткани с поражением кожи и суставов. Популяционная частота — 1:100 тысяч.
Синдром Ретта — психоневрологическое наследственное заболевание, встречается почти исключительно у девочек с частотой 1:10–1:15 тысяч. Следующая по частоте после синдрома Дауна специфическая причина тяжелой умственной отсталости у девочек. 33 – С-м Марфана. Тип наследования, патогенез, диагностика, клинические проявления, лечение.
Тип наследования: А-Д Патогенез: наличие пораженной соединительной ткани во всех внутренних структурах организма. Системная соединительнотканная недостаточность проявляется признаками поражения скелета, сердца и сосудов, глаз, кожи, ЦНС, легких. Подобные патологические изменения формируются внутриутробно у плода. Симптоматика синдрома варьируется от стертых форм до процессов, несовместимых с жизнью. Больные имеют диспропорциональные конечности, рост выше среднего, вытянутые пальцы, гипермобильные суставы, худощавое тело, готическое небо, неправильный прикус, глубоко расположенные глаза. Они страдают гигантизмом, миопией, эктопией хрусталика, изменением формы грудины, кифосколиозом. Клинические признаки синдрома обусловлены гиперрастяжимостью тканей. Клиническая картина:
Основные диагностические методики:
Лечение: Синдром Марфана неизлечим. Этиотропной терапии недуга не существует, ведь невозможно заменить гены ребенка. Больным проводят симптоматическую терапию, целью которой является облегчение общего состояния, устранение симптомов и предупреждение тяжелых осложнений. Диагностика: Лабораторная диагностика При синдроме Вольфа-Хиршхорна используют следующие лабораторные анализы: · Цитогенетический анализ; · Методика FISH Визуальная диагностика · Рентгенография · УЗИ сердца, почек · МРТ и КТ Клинические проявления: У больных детей наблюдаются микроцефалия, асимметричный череп, гипертелоризм, эпикант, косо расположенные глазные щели, птоз, нистагм, колобома радужки. Отмечается небольшой рот с опущенными углами, расщелины верхней губы и/или неба. Ушные раковины крупные, низко расположенные, нередко оттопыренные, шея короткая и тонкая, туловище вытянутое, конечности тонкие, пальцы длинные. Лечение: Симптоматическое. При пороках развития костно-мышечного аппарата, глаз, сердца, нарушениях слуха выполняют хирургическое лечение. Синдром «кошачьего крика» Тип мутации: делеция короткого плеча 5 хромосомы (5p-) Патогенез: У многих пациентов наблюдаются врожденные пороки сердца (открытый артериальный поток, тетрада Фалло), пороки развития почек (гидронефроз, подковообразная почка), крипторхизм, гипоспадия. Диагностика: 1. Бхскрининги 2. УЗИ 3. Инвизионныеметоды(амниоцентез, биопсия ворсин хориона или кордоцентез) После рождения: Осмотр и цитогенетическое исследование. Клинические проявления: Преобладание лицевой части черепа над мозговой, лунообразное лицо, гипертелоризмом, антимонголоидный разрез глаз, эпикант, деформация ушных раковин, короткая шея с крыловидными складками. Лечение: Специфического лечения не существует. Для стимуля
|
|||||||||||||
|
|
Последнее изменение этой страницы: 2021-01-08; просмотров: 359; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.8.172 (0.021 с.) |



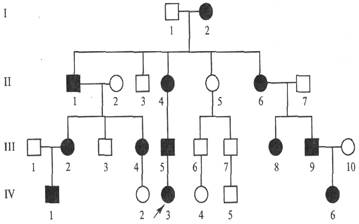 Родословная с Х-сцепленным доминантным типом наследования заболевания (витамин-Д-резистентный рахит)
Родословная с Х-сцепленным доминантным типом наследования заболевания (витамин-Д-резистентный рахит)