
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Типы окислительно-восстановительных реакцийСодержание книги
Поиск на нашем сайте
вления степени окисления изменяют атомы, входящие в состав различных соединений:
возможно и обратное диспропорционирование:
49. ГЕТЕРОГЕННЫЕ РЕАКЦИИ, хим. р-ции с участием в-в, находящихся в разл. фазах и составляющих в совокупности гетерог. систему. Типичные гетерогенные реакции: термич. разложение солей с образованием газообразных и твердых продуктов (напр., СаСО3 -> СаО + СО2), восстановление ожсидов металлов водородом или углеродом (напр., РЬО + С -> Рb + СО), растворение металлов в к-тах (напр., Zn + + H2SO4 -> ZnSO4 + Н2), взаимод. твердых реагентов (А12О3 + NiO -> NiAl2O4). В особый класс выделяют гетерогенно-каталитич. р-ции, протекающие на пов-сти катализатора; при этом реагенты и продукты могут и не находиться в разных фазах. Напр., при р-ции N2 + + ЗН2 -> 2NH3, протекающей на пов-сти железного кат., реагенты и продукт р-ции находятся в газовой фазе и образуют гомог. систему. Особенности гетерогенных реакций обусловлены участием в них конденсированных фаз. Это затрудняет перемешивание и транспортреагентов и продуктов; возможна активация молекул реагентов на пов-сти раздела фаз. Кинетика любой гетерогенной реакцииопределяется как скоростью самого хим. превращения, так и процессами переноса (диффузией), необходимыми для восполнения расхода реагирующих в-в и удаления из реакц. зоны продуктов р-ции. В отсутствие диффузионных затруднений скорость гетерогеннойреакции пропорциональна размерам реакц. зоны; т. наз. удельная скорость р-ции, рассчитанная на единицу пов-сти (или объема) реакц. зоны, не изменяется во времени; для простых (одностадийных) р-ций она м.б. определена на основе действующих масс закона. Этот закон не выполняется, если диффузия в-в протекает медленнее, чем хим. р-ция; в этом случае наблюдаемая скорость гетерогенной реакции описывается ур-ниями диффузионной кинетики (см. Макрокинетика). При гетерогенных реакциях с участием одного или неск. твердых реагентов часто образуются твердофазные продукты. Такие р-ции, как правило, локализованы на пов-сти раздела фаз или в поверхностном слое и обычно протекают нестационарно. Они характеризуютсяпериодом индукции, в течение к-рого возникают зародыши (ядра) новой фазы. Их образование связано с перестройкой атомной структуры твердого реагента и требует затраты энергии. Поэтому такие гетерогенные реакции чувствительны ко всем нарушениям структуры, облегчающим образование зародышей, и м. б. активированы термич., радиац., мех. и др. воздействиями, увеличивающимиконцентрацию дефектов, в первую очередь плотность дислокаций (см. Дефекты в кристаллах). Кинетич. ур-ние р-ции в этом случае отражает изменение во времени не только концентраций реагирующих в-в, но и пов-сти раздела твердых фаз реагента и продукта: по мере роста зародышей пов-сть раздела увеличивается и скорость р-ции сначала возрастает, затем проходит через максимум и снижается вследствие соприкосновения растущих зародышей и образования сплошного слоя твердого продукта (подробнее см.Топохимические реакции). В природе гетерогенные реакции входят в комплекс процессов, приводящих к образованию осадочных пород и выветриванию. В хим. технологии гетерогенные реакции газа с жидкостью (окисление воздухом, кислородом, озоном; хлорирование и др.) обычно проводят при интенсивном перемешивании спец. мех. устройствами или самим газом (в т. наз. барботажном реакторе). Р-ции термич. разложения составляют основу фотографич. процесса, р-ции между газами или жидкостями и твердыми в-вами-основу обжига,восстановления и окисления металлов, горения, произ-ва твердых катализаторов, выщелачивания, экстракции и др. Часто сочетаются гетерогенные реакции в трех-и многофазных системах, напр. хлорирование твердых оксидов металлов газообразным хлором в присут. твердого углеродсодержащего восстановителя. Важная область использования гетерогенных реакций-получение тонких поверхностных слоев и покрытий при взаимод. твердого тела с жидкостью. При низких т-рах диффузия в глубь твердого материала протекает медленно, что позволяет получать стабильные тонкие поверхностные слои, а в отдельных случаях-двухмерные фазы, толщина к-рых по порядку величины близка к параметру кристаллич. решетки. Иногда стабильные поверхностные слои образуются самопроизвольно; таковы защитные оксидные пленки на металлах, препятствующие дальнейшему окислению (см. Газовая коррозия). ЭЛЕКТРОДНЫЙ ПОТЕНЦИАЛ, разность электростатич. потенциалов между электродом и находящимся с ним в контакте электролитом. Возникновение электродного потенциала обусловлено пространств. разделением зарядов противоположного знака на границе раздела фаз и образованием двойного электрического слоя. На границе между металлич. электродом и р-ром электролита пространств. разделение зарядов связано со след. явлениями: переносом ионов из металла в р-р в ходе установления электрохим. равновесия, кулоновской адсорбцией ионов из р-ра на пов-сть металла, смещением электронного газа за пределы положительно заряженного ионного остова кристаллич. решетки, специфич. (некулоновской) адсорбцией ионов или полярных молекул р-рителя на электроде и др. Последние два явления приводят к тому, что электродный потенциал не равен нулю даже при условиях, когда заряд пов-сти металларавен нулю (см. Потенциал нулевого заряда).
где vi - стехиометрич. коэф. участника р-ции, причем для исходных в-в это отрицат. величина, а для продуктов р-ции -положительная. 50. Химические источники тока, устройства, вырабатывающие электрическую энергию за счёт прямого преобразования химической энергии окислительно-восстановительных реакций. Первые химические источники тока созданы в 19 в. (Вольтов столб, 1800; элемент Даниела — Якоби, 1836; Лекланше элемент, 1865, и др.). До 60-х гг. 19 в. химические источники тока были единственными источниками электроэнергии для питания электрических приборов и для лабораторных исследований. Основу химических источников тока составляют два электрода (один — содержащий окислитель, другой — восстановитель), контактирующие с электролитом. Междуэлектродами устанавливается разность потенциалов — электродвижущая сила (эдс), соответствующая свободной энергииокислительно-восстановительной реакции. Действие химических источников тока основано на протекании при замкнутой внешней цепи пространственно разделённых процессов: на отрицательном электроде восстановитель окисляется, образующиеся свободныеэлектроны переходят по внешней цепи (создавая разрядный ток) к положительному электроду, где участвуют в реакции восстановленияокислителя. В зависимости от эксплуатационных особенностей и от электрохимической системы (совокупности реагентов и электролита) химические источники тока делятся на гальванические элементы (обычно называются просто элементами), которые, как правило, после израсходования реагентов (после разрядки) становятся неработоспособными, и аккумуляторы, в которых реагенты регенерируются при зарядке — пропускании тока от внешнего источника (см. Зарядное устройство). Такое деление условно, т.к. некоторые элементы могут быть частично заряжены. К важным и перспективным химическим источникам тока относятся топливные элементы (электрохимические генераторы), способные длительно непрерывно работать за счёт постоянного подвода к электродам новых порций реагентов и отвода продуктов реакции. Конструкция резервных химических источников тока позволяет сохранять их в неактивном состоянии 10—15 лет (см. также Источники тока). С начала 20 в. производство химических источников тока непрерывно расширяется в связи с развитием автомобильного транспорта, электротехники, растущим использованием радиоэлектронной и др. аппаратуры с автономным питанием. Промышленность выпускает химические источники тока, в которых преимущественно используются окислители PbO2, NiOOH, MnO2 и др., восстановителями служат Pb, Cd. Zn и др. металлы, а электролитами — водные растворы щелочей, кислот или солей (см., например, Свинцовый аккумулятор). Основные характеристики ряда химических источников тока приведены в табл. Лучшие характеристики имеют разрабатываемые химические источники тока на основе более активных электрохимических систем. Так, в неводных электролитах (органическихрастворителях, расплавах солей или твёрдых соединениях с ионной проводимостью) в качестве восстановителей можно применятьщелочные металлы (см. также Расплавные источники тока). Топливные элементы позволяют использовать энергоёмкие жидкие или газообразные реагенты. 51. Электро́лиз — физико-химический процесс, состоящий в выделении на электродах составных частей растворённых веществ или других веществ, являющихся результатом вторичных реакций на электродах, который возникает при прохождении электрического тока через раствор либо расплав электролита. Электролиз - это ещё один способ получения чистых металлов и неметаллов. Кроме того, электролиз можно провести и в домашних условиях. Нужен источник тока, два электрода (какие электроды бывают и какой в каком случае брать - расссказано дальше) и, конечно, электролит. Электролит - это раствор, который проводит электрический ток. Различают электролиз растворов и электролиз расплавов. Оба эти процесса существенно отливчаются друг от друга. Отличие - в наличии растворителя. При электрролизе растворов кроме ионов самого вещества в процессе учавствуют ионы растворителя. При электролизе расплавов - только ионы самого вещества. Применение электролиза для обработки поверхностей включает как катодные процессы гальванотехники (в машиностроении, приборостроении, авиационной, электротехнической, электронной промышленности), так и анодные процессы полировки, травления, размерной анодно-механической обработки, оксидирования (анодирования) металлических изделий (см. также Электрофизические и электрохимические методы обработки). Путём электролиза в контролируемых условиях осуществляют защиту от коррозии металлических сооружений и конструкций (анодная и катодная защита). 52. ФАРАДЕЯ ЗАКОНЫ, основные законы электролиза, отражающие общий закон сохранения в-ва в условиях протекания злектрохим. р-ции. Установлены M. Фарадеем в 1833-34. Согласно 1-му закону, масса в-ва т, прореагировавшего в процессе электролиза, прямо пропорциональна силе тока I и времени электролиза t, т. е. кол-ву пропущенного электричества Q = It (предполагается, что I не зависит от t; в противном случае масса т пропорциональна
где M - мол. м. в-ва, участвующего в электролизе, z - число элементарных зарядов, соответствующее превращению одной молекулыэтого в-ва, 1/F- коэф. пропорциональности, общий для всех в-в, F - Фарадея постоянная, равная 96484,56 Кл/моль. Фарадея законы относятся к числу строгих законов, но в ряде случаев могут наблюдаться кажущиеся отклонения от них, вызываемые след. причинами: 1) в нестационарных условиях электролиза часть электричества затрачивается на заряжение двойного электрического слоя; 2) если электролит обладает электронной проводимостью (напр., р-р металлич. Na в жидком аммиаке), то часть тока черезэлектролит переносят электроны, а не ионы, и соответствующее кол-во электричества не участвует в процессе электролиза; 3) наряду с основным процессом электролиза, напр, образованием металлич. Zn по р-ции Zn2+ + 2е Фарадея законы сыграли важную роль в понимании природы хим. связи и развития атомно-молекулярной теории. Их используют при выводе всех ур-ний, описывающих электрохим. превращения B-B на границах раздела проводников 1-го и 2-го рода (см.Электрохимическая кинетика). Практич. применение Фарадея законы находят в кулонометрии, а также при определении выхода р-ции по току, т.е. отношения теоретич. кол-ва электричества, рассчитанного на основе Фарадея законов, к кол-ву электричества, реально затраченному на получение данного в-ва в процессе электролиза. Поляризация электрохимическая, отклонение электродного потенциала Е от стационарного потенциала Ест, который электродприобретает в отсутствие внешнего тока. Поляризация электрохимическая измеряется в вольтах (милливольтах). Если отклонение отрицательно (вызвано подводом электронов, которые должны расходоваться в реакциях, идущих в катодном направлении), тополяризацию электрохимическую называют катодной; при противоположном направлении тока — анодной. Графики функциональной связи между поляризацией электрохимической и плотностью тока i называют соответственно катодными и анодными поляризационными кривыми и широко используют при описании и исследовании электрохимических и коррозионных процессов. В общем случае связь между i и поляризацией электрохимической криволинейна, однако в интервале отклонений ± 10—15 мв от Естона, как правило, прямолинейна. Угловой коэффициент этого участка (т. е. отношение приращения поляризации электрохимической к приращению i) имеет размерность сопротивления единицы поверхности (омсм2) и называется поляризационным сопротивлениемэлектрода Rп. Электроды с большим Rп называются сильнополяризуемыми, т.к. уже при очень малых i их потенциалы сильно отклоняются от Ест. Электроды с малым Rп — слабополяризуемые. Существует обратная пропорциональность между Rп и интенсивностью того обмена электрическими зарядами, который происходит между электродом и электролитом при Ест. На коррелирующем электроде эта интенсивность обычно совпадает с плотностью коррозионного тока, и потому измерение Rп иногда используют для определения скорости электрохимической коррозии. Если на электроде возможна лишь одна электродная реакция, то Ест совпадает с равновесным потенциалом Ер этой реакции, поляризация электрохимическая — с её перенапряжением, a Rпоказывается обратно пропорциональным равновесному току обмена. Термином «концентрационная поляризация» обозначают те изменения Е, которые связаны с замедленным переносом исходных или конечных компонентов протекающей на электроде реакции. В зоне реакции концентрация первых (сисх) понижается, а вторых (скон) — увеличивается. Это повышает тенденцию реакции протекать в обратном направлении, что и должно компенсироваться приложением дополнительной разности потенциалов. Последняя особенно резко растет, когда скорость реакции достигает предельно возможной скорости диффузионных потоков, так что либо сисх снижается практически до 0, либо конечные продукты кристаллизуются, закрывая электродную поверхность. Эту предельную диффузионную плотность тока можно повысить, улучшив массоперенос, например, путёмперемешивания. Вместо термина «концентрационная поляризация» также пользуются термином «концентрационноеперенапряжение», т.к. обозначаемое им отклонение Е должно фактически отсчитываться не от Ест, а от Ер соответствующей индивидуальной реакции. Явления поляризации электрохимической могут быть и вредны, и полезны. Например, при электролизе они повышают расход электроэнергии, а при работе гальванического элемента понижают отдачу электроэнергии; зато при коррозии могут вести к торможению нежелательных процессов. См. также ст. Пассивирование. Перенапряжение электрохимическое, отклонение электродного потенциала от его равновесного (по отношению к приэлектродному составу раствора) термодинамического значения при поляризации электрода внешним током. При заметном удалении от равновесияперенапряжение () и плотность поляризующего тока (i) обычно связаны соотношением = а + b lg i (уравнение Тафеля), где а и b — эмпирические постоянные. Перенапряжение зависит от температуры, природы электродного материала и состава раствора. Перенапряжение необходимо для ускорения нужной электродной реакции. Если скорость электродной реакции в целом определяется скоростью собственно электрохимической стадии, связанной с переносом заряда, то перенапряжение усиливает электрическое поле, действующее на разряжающиеся частицы, благодаря чему снижается энергия активации разряда. Поскольку электрическое поле в значительной степени обусловлено строением двойного электрического слоя, перенапряжение оказывается зависящим отконцентрации постороннего электролита и адсорбирующихся веществ, влияющих на распределение потенциала в двойном слое. На повышении перенапряжения основано действие многих ингибиторов коррозии металлов (см. Ингибиторы химические), что является одной из положительных сторон перенапряжения. В то же время перенапряжение в промышленном электролизе, неизбежно связанное с дополнительным расходом энергии, приводит к увеличению себестоимости продукции. 53. Коррозия металлов Коррозия металлов — разрушение металлов вследствие химического или электрохимического взаимодействия их с коррозионной средой.[2] Для процесса коррозии следует применять термин «коррозионный процесс», а для результата процесса — «коррозионное разрушение». Образование гальванических пар с пользой применяют для создания батарей и аккумуляторов. С другой стороны, образование такой пары приводит к неблагоприятному процессу, жертвой которого становится целый ряд металлов, — коррозии. Под коррозией понимают происходящее на поверхности электрохимическое или химическое разрушение металлического материала. Наиболее часто при коррозии металл окисляется с образованием ионов металла, которые при дальнейших превращениях дают различные продукты коррозии. Коррозия может быть вызвана как химическим, так и электрохимическим процессом. Соответственно, различают химическую и электрохимическую коррозию металлов. [ править ] Типы коррозии [ править ] Электрохимическая коррозия Разрушение металла под воздействием возникающих в коррозионной среде гальванических элементов называют электрохимической коррозией. Не следует путать с электрохимической коррозией коррозию однородного материала, например, ржавление железа или т. п. При электрохимической коррозии (наиболее частая форма коррозии) всегда требуется наличие электролита (Конденсат, дождевая вода и т. д.), с которым соприкасаются электроды — либо различные элементы структуры материала, либо два различных соприкасающихся материала с различающимися окислительно-восстановительными потенциалами. Если в воде растворены ионы солей, кислот, или т. п., электропроводность её повышается, и скорость процесса увеличивается.
Коррозионный элемент При соприкосновении двух металлов с различными окислительно-восстановительными потенциалами и погружении их в раствор электролита, например, дождевой воды с растворенным углекислым газом CO2, образуется гальванический элемент, так называемый коррозионный элемент. Он представляет собой не что иное, как замкнутую гальваническую ячейку. В ней происходит медленное растворение металлического материала с более низким окислительно-восстановительным потенциалом; второй электрод в паре, как правило, не корродирует. Этот вид коррозии особо присущ металлам с высокими отрицательными потенциалами. Так, совсем небольшого количества примеси на поверхности металла с большим редокспотенциалом уже достаточно для возникновения коррозионного элемента. Особо подвержены риску места соприкосновения металлов с различными потенциалами, например, сварочные швы или заклёпки. Если растворяющийся электрод коррозионно-стоек, процесс коррозии замедляется. На этом основана, например, защита железных изделий от коррозии путём оцинковки — цинк имеет более отрицательный потенциал, чем железо, поэтому в такой паре железо восстанавливается, а цинк должен корродировать. Однако в связи с образованием на поверхности цинка оксидной плёнки процесс коррозии сильно замедляется.
|
||
|
|
Последнее изменение этой страницы: 2016-04-07; просмотров: 427; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.33 (0.016 с.) |








 CuCl2|Cu|Pt (две штриховые черты означают, что диффузионный потенциал на фанице НС1 и СuС12 устранен).
CuCl2|Cu|Pt (две штриховые черты означают, что диффузионный потенциал на фанице НС1 и СuС12 устранен).  и константой равновесия Кр электрохим. р-ции ур-нием:
и константой равновесия Кр электрохим. р-ции ур-нием:  , где F - число Фарадея; п - число электронов, участвующих в р-ции; R - газовая постоянная; Т - абс. т-ра. Значения E° электрохим. систем по отношению к водородному электроду и протекающие на электродах р-ции сведены в спец. таблицы (подробнее см. Стандартный потенциал).
, где F - число Фарадея; п - число электронов, участвующих в р-ции; R - газовая постоянная; Т - абс. т-ра. Значения E° электрохим. систем по отношению к водородному электроду и протекающие на электродах р-ции сведены в спец. таблицы (подробнее см. Стандартный потенциал). 
 где t1 и t2 - моменты включения и выключения тока). Согласно 2-му закону, для разных электродных процессов при одинаковом кол-ве пропущенного электричества Q массы прореагировавших в-в относятся друг к другу так же, как эквиваленты химические этих в-в. Оба Фарадея закона объединяются одним ур-нием:
где t1 и t2 - моменты включения и выключения тока). Согласно 2-му закону, для разных электродных процессов при одинаковом кол-ве пропущенного электричества Q массы прореагировавших в-в относятся друг к другу так же, как эквиваленты химические этих в-в. Оба Фарадея закона объединяются одним ур-нием: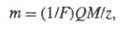
 Zn, часть тока может затрачиваться на протекание параллельных электрохим. р-ций, напр.: 2H3O+ + 2е = H2 + 2H2O; O2 + 4е + 4H3O+ = 6H2O. Системы, в к-рых полностью исключены указанные причины кажущихся отклонений от Фарадея законов, получили назв. кулонометров; их использование позволяет по кол-ву образовавшихся продуктов электролиза точно определить кол-во пропущенного электричества. В кулонометрах обычно применяют электрохим. р-ции Ag+ + е = Ag или 3I- = I3- + 2е.
Zn, часть тока может затрачиваться на протекание параллельных электрохим. р-ций, напр.: 2H3O+ + 2е = H2 + 2H2O; O2 + 4е + 4H3O+ = 6H2O. Системы, в к-рых полностью исключены указанные причины кажущихся отклонений от Фарадея законов, получили назв. кулонометров; их использование позволяет по кол-ву образовавшихся продуктов электролиза точно определить кол-во пропущенного электричества. В кулонометрах обычно применяют электрохим. р-ции Ag+ + е = Ag или 3I- = I3- + 2е.



