
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Факторы, влияющие на всасывание
1. Лекарственная форма. Определяет скорость высвобождения лекарственного вещества в месте введения. 2. Растворимость в воде. Лекарства, которые вводят в организм в виде таблеток, драже, порошков, других твердых лекарственных форм, или, например, суспензий, аэрозолей порошков, должны раствориться в водной биофазе, прежде чем высвобождающиеся при этом молекулы лекарственных веществ абсорбируются. Очевидно, что лекарственные вещества, которые вводят в виде водных растворов, абсорбируется быстрее, чем при их введении в виде твердых лекарственных форм, взвесей или растворов в масле. Для плохо растворимых в воде лекарств (как, например, ацетилсалициловая кислота) скорость растворения управляет скоростью абсорбции. 3. Концентрация. И липидная диффузия, и фильтрация зависят от концентрационного градиента. Чем выше концентрация лекарственного вещества в месте абсорбции, тем выше скорость всасывания. Создание высокой концентрации лекарства в месте введения приводит к ускорению абсорбции. Лекарства, вводимые в виде концентрированных растворов, абсорбируются быстрее. 4. Площадь абсорбирующей поверхности. Абсорбция является поверхностным феноменом и в соответствии с законом Фика, чем большая площадь абсорбирующей поверхности, тем быстрее абсорбция. 5. Кровоснабжение абсорбирующей поверхности. Ток крови удаляет молекулы лекарства с места абсорбции, что способствует поддержанию концентрационного градиента. 6. Путь введения. Каждый имеет свои особенности. - пероральный. Основным барьером на пути абсорбируемых веществ, является слизистая оболочка, которая состоит из плотно прилегающих друг к другу клеток, что делает ее непроницаемой для нерастворимых в липидах ионизированных молекул лекарств. Неионизирующиеся, растворимые в липидах вещества, такие, как, например, этанол, легко абсорбируются в желудке, так же как и в кишечнике. Лекарства, являющиеся слабыми кислотами (например, салицилаты, барбитураты), находящиеся в желудочном соке преимущественно в неионизированном состоянии, могут абсорбироваться в желудке, в то время как лекарства-слабые основания (например, морфин, хинидин), которые в желудке более ионизированы, абсорбируются, только в 12-перстной кишке. Однако, даже лекарства-кислоты абсорбируются в желудке медленно, потому что слизистая желудка покрыта слизью, а площадь всасывания небольшая. Вследствие этого более быстрая эвакуация содержимого желудка в 12-перстную кишку в целом ускоряет абсорбцию.
На абсорбцию лекарств из ЖКТ влияет присутствие пищи. Большинство лекарств всасывается лучше, если их принимать на пустой желудок. Пища абсорбирует лекарство, понижая, таким образом, концентрацию его свободных молекул. Некоторые лекарственные вещества образуют плохо абсорбирующиеся комплексы с компонентами пищи (например, тетрациклины с кальцием, присутствующем в молоке, йогурте). Абсорбция лекарств может быть изменена другими лекарствами, назначаемыми одновременно: вследствие непосредственного взаимодействия между ними, которое приводит к образованию не всасывающихся комплексов (например, тетрациклинов с лекарственными средствами железа или фторхинолонов с антацидами), из-за изменения перистальтики (например, под влиянием антихолинэстеразных средств, опиоидных анальгетиков, веществ с атропиноподобным действием, прокинетиков), или повреждения слизистой, что приводит к развитию синдрома мальабсорбции (метатрексат, неомицин). - подкожное и внутримышечное введение. При этих способах введения лекарство доставляется непосредственно в межклеточное пространство, окружающее капилляры. Растворимые в липидах вещества легко проникают через эндотелий капилляров. Поры в капиллярах не пряпятствуют абсорбции даже нерастворимых в липидах веществ и ионизированных молекул. Очень большие молекулы абсорбируются в лимфатические сосуды. Поэтому многие лекарственные вещества, которые не абсорбируются при пероральном приеме, абсорбируются при парентеральном введении. Абсорбция с подкожных депо более медленная, чем с мест внутримышечного введения, но и с того и другого более быстрая, более постоянная и более предсказуемая, чем абсорбция при приеме внутрь. Прикладывание тепла или мышечные упражнения ускоряют абсорбцию из-за увеличения кровотока, в то время как вазоконстрикторы, добавленные к растворам вводимых веществ, наоборот, задерживают абсорбцию.
-местное применение лекарств. Системная абсорбция после местной аппликации лекарства определяется, прежде всего, его растворимостью в липидах. Коньюнктива, слизистая оболочка полости рта, прямой кишки, вагины является проницаемой только для растворимых в липидах неионизированных веществ. Лишь немногие лекарственные вещества проникают через неповрежденную кожу (нитроглицерин, фентанил, никотин, эстрадиол, тестостерон, кортикостероиды). Абсорбции через кожу способствуют втирание лекарств с маслом, плотное укутывание (увеличивает потоотделение и таким образом гидратацию кожи), повреждение кожи (потертые или ожоговые поверхности).
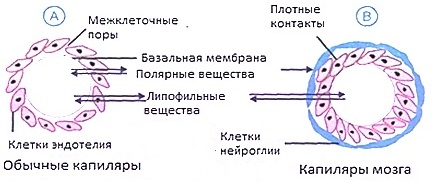
Транспорт и распределение лекарств в организме. Связывание лекарственных веществ белками плазмы крови. Транспорт через гистогематические барьеры. Депонирование лекарств в тканях. Обьем распределения. Распределение лекарств – это распространение лекарственных веществ по органам и тканям после их попадания в системный кровоток. Зависит главным образом от природы лекарства, интенсивности кровотока в тканях, проницаемости гистогематических барьеров, а также связывания молекул лекарства с белками плазмы крови и в тканях. 1. Природа лекарств. Определяет прежде всего возможность переноса через биологические барьеры. Наибольшее значение имеют размеры молекул и их полярность, степень ионизации. Большинство гидрофильных лекарственных веществ не проникают в клетки и распределяются в основном в плазме крови и интерстициальной жидкости. Липофильные лекарства относительно легко проникают через гистогематические барьеры, диффундируют в клетки и распределяются в организме более равномерно. 2. Кровоток. Приток крови обеспечивает доставку лекарства в ткани и таким образом влияет на скорость захвата лекарственного вещества тканями. В результате в хорошо перфузируемых тканях (например, мозг, сердце, почки) большие тканевые концентрации создаются раньше, чем в плохо перфузируемых (например, жировой, костной). Если при этом лекарство быстро элиминируется, то его концентрация в плохо перфузируемых тканях может никогда существенно и не повысится. 3. Связывание лекарств с белками плазмы. Затрудняет диффузию лекарства в периферические ткани. Это происходит вследствие того, что диффундировать через поры в капиллярах могут только свободные молекулы. Самой большой фракцией белков в плазме крови является альбумин. Более высокое сродство альбумин проявляет к гидрофобным веществам и лекарствам, являющимся слабыми кислотами. Связывание лекарственных веществ с белками плазмы крови процесс обратимый и не является специфичным. Лекарственные вещества, при их одновременном назначении могут конкурировать за места связывания на белковых молекулах и вытеснять друг друга. Уменьшение связывания лекарственного вещества с белками плазмы может привести к существенному увеличению фракции его свободных молекул в крови и явиться причиной чрезмерного усиления фармакологического действия лекарства. 4. Гистогематические барьеры. Это барьеры между кровью и тканями, образованные стенкой капилляров. Не одинаковы в различных органах и тканях. Например, в ЦНС он наименее проницаем, так как в его образовании принимают участие еще и клетки нейроглии:
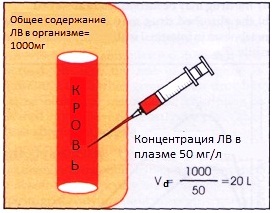
В целом перенос лекарственных веществ через подобного рода барьеры подчиняется закономерностям, характерным для механизмов абсорбции, описанным ранее, и зависит от природы вещества: лучше переносятся неполярные липофильные вещества, хуже – полярные, гидрофильные. Многие лекарства в физиологических условиях не проникают через гистогематические барьеры, например, маннитол, высокомолекулярные декстраны (полиглюкин). Через гематоэнцефалический барьер не проникают нейромедиаторы и плохо проходят полярные соединения. 5. Связывание лекарства в тканях. Способствует переходу лекарства из крови и накоплению его в тканях, так как связывание понижает концентрацию свободных молекул лекарственного вещества непосредственно в периваскулярном пространстве и таким образом поддерживает высоким градиент способных к диффузии (несвязанных) молекул вещества. Это может приводить к значительному накоплению (депонированию) лекарства в периферических тканях. При обратимом связывании лекарственное вещество может постепенно высвобождаться из депо и, при понижении его концентрации в крови, снова подвергаться распределению. О распределении лекарств принято судить по объему распределения. Объем распределения (Vd - от Volume of distribution) связывает количество лекарства в организме с его концентрацией в плазме в соответствии со следующим уравнением:
Количественно равен условному объему в котором следовало бы распределить все лекарство, содержащееся в организме, чтобы его концентрация в этом объеме была равна таковой в плазме. Если лекарство имеет очень большой объем распределения, значительно превышающий физический объем тела,это означает, что лекарственное вещество в основном находится в периферических тканях в связанном состоянии. Такие лекарства не могут быть эффективно удалены из организма с помощью гемодиализа. С другой стороны, вещества, которые полностью остаются в плазме, будут иметь объем распределения равный объему плазмы (приблизительно 3 ‒ 4 литра), что характерно для высокомолекулярных соединений, не проникающих в клетки крови и через поры в капиллярах (например, гепарин). Если Vd равен 15 л (суммарный объем плазмы крови и интерстициальной жидкости) лекарство преимущественно распределено внеклеточно, что характерно для гидрофильных веществ, таких, например, как аминогликозидные антибиотики.
При величине объема распределения порядка 40 л (объем всех жидкостей в организме) лекарство вероятнее всего находится как во внеклеточной, так и внутриклеточной жидкостях, то есть проникает через клеточные мембраны, что характерно для распределения липофильных неполярных веществ. Величина объема распределения играет важную роль в оценке элиминации лекарственных веществ из организма (при прочих равных условиях вещество с большим Vd будет элиминироваться медленнее и наоборот), а также учитывается при определении нагрузочной дозы: нагрузочная доза = желаемая (или целевая) концентрация лекарственного вещества × Vd.
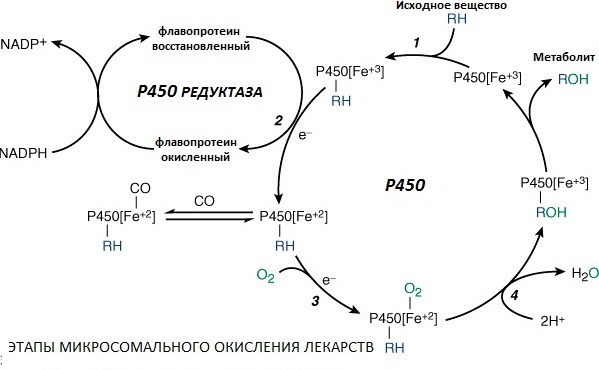
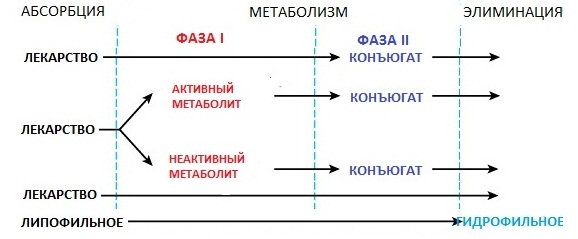
Биотрансформация лекарственных веществ в организме. Несинтетические и синтетические реакции метаболизма лекарств. Роль микросомальных ферментов печени. Эффект первого прохождения. Внепеченочный метаболизм лекарственных веществ. Понятие о «пролекарствах». Индивидуальные различия в скорости инактивации лекарств и причины их обусловливающие. Попав в организм, большинство лекарственных веществ, будучи ксенобиотиками с липофильными свойствами, вовлекаются в метаболизм и подвергаются в организме химическим превращениям. Различают 2 вида таких превращений лекарственных веществ: · несинтетические (или метаболической трансформации, или реакции фазы I) · синтетические (или конъюгации, или фазы II). Несинтетические реакции Это превращение веществ за счет окисления, восстановления, гидролиза. Большая часть несинтетических реакций катализируется микросомальными ферментами печени.
Микросомальные ферменты сосредоточены внутри клеток и связаны с мембранами гладкого эндоплазматического ретикулума. К ним относят флавопротеин: НАДФ-Н-цитохром Р-450 редуктазу и гемопротеин: цитохром Р-450. Идентифицировано более 100 изоформ цитохрома Р-450 (Cytochrome P-450, CYP), различающихся по своей аффинности для различных субстратов. Наиболее важными для людей являются следующие изоэнзимы цитохрома Р–450: CYP 3A4/5. Обеспечивает биотрансформацию наибольшего количества (50%) лекарств. В дополнение к печени, эта изоформа представлена также в кишечнике (ответственна за метаболизм первичного прохождения в этом органе) и почках. CYP 2D6. Изоформа с участием которой метаболизируется приблизительно 20% лекарств, включая трициклические антидепрессанты, селективные ингибиторы обратного захвата серотонина, многие нейролептики, антиаритмические средства, β-адреноблокаторы и опиоиды. CYP 2C8/9. Важен для биотрансформации 15 лекарственных веществ, включая фенитоин и варфарин, относящихся к лекарственным средствам с узкой терапевтической широтой. CYP 2E1. Катализирует образование минорных метаболитов некоторых лекарств, в том числе N-ацетилбензохинонимина из парацетамола; индукция этого изофермента имеет место при хроническом алкоголизме. Часть несинтетических реакций не требует участия микросомальных ферментов и может осуществляться как в печени с участием митохондриальных или цитозольных ферментов (адреналин, этанол, меркаптопурин), так и в других органах и тканях (кишечнике, почках, коже, легких, мышцах, крови, межклеточной жидкости), например, гидролиз эфиров, амидов, полипептидов.
Синтетические реакции Это реакции, в основе которых лежит связывание (конъюгация) метаболитов лекарств или, реже, неизмененных лекарственных веществ с глюкуронидной, ацетильной, сульфатной, метильной группами, а также глютатионом или глицином. Все виды конъюгации (за исключением глюкуронидной) катализируются немикросомальными ферментами. Конъюгация является основным видом метаболизма карбоновых кислот, спиртов, фенолов. Путем конъюгации из организма элиминируются эстрогены, глюкокортикоиды, прогестерон, опиоиды, салицилаты, барбитураты, хлорамфеникол. В большинстве случаев лекарственного метаболизма несинтетические реакции предшествуют реакциям конъюгации. В результате превращений лекарственное вещество, как правило, становится более растворимым в воде, что позволяет ускорить его выведение из организма:
Фармакологическая активность в результате биотрансформации утрачивается. Таким образом, метаболизм лекарств является одним из механизмов их элиминирования. Хотя важны и другие аспекты лекарственного метаболизма.
Скорость биотрансформации лекарств может заметно отличаться у разных пациентов. Эта вариабельность в основном является следствием генетических различий, воздействием на организм других веществ, наличием сопутствующих заболеваний, половыми и возрастными особенностями. 1. Генетические факторы. Например, генетический дефект активности псевдохолинестеразы, который встречается приблизительно у 1 человека из 2500, приводит к выраженному замедлению гидролиза сукцинилхолина (и сходных с ним эфиров) и резкому увеличению продолжительности нервно-мышечного паралича, вызванного этим веществом. Аналогичные фармакогенетические влияния выявлены в отношении ацетилирования изониазида (противотуберкулезного средства), прокаинамида (противоаритмического средства), окисления некоторых трициклических антидепрессантов. 2. Взаимодействие лекарств в процессе метаболизма. Одновременное назначение некоторых лекарств может приводить к заметному изменению лекарственного метаболизма из-за вызываемой ими индукции или ингибирования ферментов печени, принимающих участие в биотрансформации лекарств. К важнейшим индукторам относятся барбитураты, фенилбутазон (нестероидное противовоспалительное средство), фенитоин (противоэпилептическое средство), рифампицин (антибиотик), гризеофульвин (противогрибковое средство) и некоторые токсические вещества, такие как бензпирен (из табачного дыма) и пестициды. Ингибиторами ферментов являются аллопуринол (урикозурическое средство), хлорамфеникол (антибиотик), циметидин (блокатор Н2 гистаминовых рецепторов), эритромицин, кларитромицин (антибиотики из группы макролидов), кетоконазол, итраконазол (противогрибковые средства). 3. Заболевания, влияющие на метаболизм лекарств. Острые или хронические заболевания, влияющие на структуру и функцию печени заметно изменяют печеночный метаболизм многих лекарств. В зависимости от тяжести такие заболевания приводят к нарушению функций печеночных ферментов, метаболизирующих лекарства или вызывают понижение запасов конъюгирующих агентов: глюкуронидов, сульфатов, глютатиона. Заболевания сердца за счет ограничения печеночного кровотока, могут нарушить кинетику лекарств, метаболизм которых лимитируется кровотоком, например, пропранолола (бета-блокатор), верапамила (блокатор кальциевых каналов), лидокаина (местный анестетик, противоаритмическое средство). 4. Возраст и пол. У человека выявлены тенденции к замедлению метаболизма при старении. Пол может быть важным для лекарств, метаболизирующихся с участием CYP2D6 (большинство β-адреноблокаторов), зкспрессия которого зависит от уровня тестостерона и выше у мужчин. У женщин, показано, выше активность CYP3A4. В эмбриональном периоде отсутствует большинство ферментов метаболизма лекарств, у новорожденных в первый месяц жизни активность этих ферментов снижена и достигает достаточного уровня лишь через 1 – 6 месяцев.
Пути выведения лекарств из организма. Механизмы почечной экскреции и факторы, влияющие на выделение веществ с мочой. Циркуляция лекарственных веществ в организме Лекарственные вещества и их метаболиты выводятся из организма через:
Выведение через почки Является основным путем экскреции лекарств из организма. Включает 3 процесса: клубочковую фильтрацию, проксимальную канальцевую секрецию и дистальную канальцевую реабсорбцию. Фильтрация. Фильтрации подвергаются практически все вещества с молекулярной массой менее 20 000 и не связанные с белками плазмы или форменными элементами крови. Скорость фильтрации определяется градиентом давления в клубочках и капсуле почечного канальца, а также количеством функционирующих клубочков. Маркером фильтрационной способности почек является клиренс креатинина, который можно определить лабораторным путем или рассчитать по специальным формулам, например Кокрофта и Голта (Cocroft D. W., Gault M. N.). Для расчета клиренса креатинина по этой формуле необходимо знать только один биохимический параметр ‒ креатинин сыворотки крови, определение которого возможно в любой лаборатории: Клиренс креатинина (мл/мин) = Для расчета клиренса у детей используется формула Шварца (Schvarz G. L.): Клиренс креатинина (мл/мин) = Секреция. Молекулы лекарств, являющихся слабыми кислотами (например, антибиотик бензилпенициллин), или основаниями (например, мочегонное средство триамтерен) выводятся активно в ионизированном состоянии с участием специальных транспортных систем эпителия проксимальных канальцев. Связывание с белками не влияет существенно на процесс секреции. Более важным является конкурентное взаимодействие, возникающее между одновременно выводимыми с помощью такого механизма, веществами, так как специфичность упомянутых транспортных систем низкая. Такой механизм лежит в основе, как полагают, замедления экскреции бензилпенициллина при его комбинированном назначении с пробеницидом. Реабсорбция. По мере продвижения выводимого вещества по канальцу его концентрация возрастает и может превысить таковую в интерстициальном пространстве и вещество может по градиенту концентраций диффундировать обратно из канальцевой мочи в кровь. Так как диффузия зависит от степени ионизации, то реабсорбцией можно управлять, изменяя pH мочи. Подкисление мочи (например, с помощью хлористого аммония) способствует выведению слабых оснований, подщелачивание (например, с помощью натрия гидрокарбоната) усиливает экскрецию слабых кислот. Это происходит благодаря повышению содержания ионизированных молекул выводимых веществ (см. уравнение Хендерсона-Хассельбаха).
|
|||||||||
|
|
Последнее изменение этой страницы: 2017-02-21; просмотров: 350; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.84.175 (0.034 с.) |
 .
.
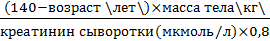 (для мужчин). Для женщин все следует умножить на 0,85.
(для мужчин). Для женщин все следует умножить на 0,85. , где К – возрастной коэффициент пересчета
, где К – возрастной коэффициент пересчета


