Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
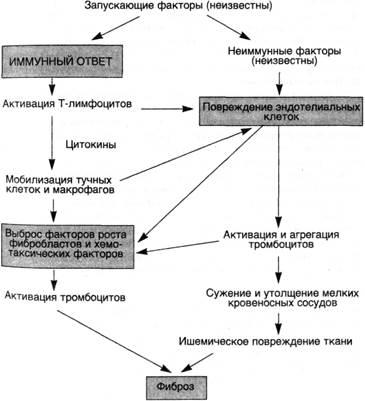
Схема 32. Патогенез прогрессирующего системного склероза (склеродермии)Содержание книги
Поиск на нашем сайте
■ Таким образом, в основе прогрессирующего системного склероза лежат различные иммунные нарушения, выраженный фиброз и изменения микроциркуляторного русла. Хотя антигены, запускающие аутоиммунный ответ, и не идентифицированы, установлено, что именно иммунологические механизмы вызывают развитие фиброза с помощью цитокинов, которые активируют фибробласты, или посредством повреждения мелких кровеносных сосудов, либо благодаря обоим механизмам. Воспалительные миопатии. Это гетерогенная группа заболеваний, характеризующихся иммунологически обусловленным воспалением скелетных мышц. К ним относятся дер-матомиозит и полимиозит, которые могут развиваться сами по себе или сочетаться с другими иммунологически обусловленными болезнями, обычно с прогрессирующим системным склерозом. Дерматомиозит характеризуется поражением кожи и скелетных мышц, встречается у детей и взрослых. Классическая сыпь при этом заболевании возникает в виде сиреневых или обесцвеченных участков на верхних веках и сопровождается периорбитальным отеком. Нередко появляются шелушащиеся эритематозные высыпания или темно-красные пятна на суставах и локтях. Мышечная слабость развивается медленно, бывает двусторонней симметричной и обычно вначале поражает проксимальные мышцы, поэтому первыми симптомами заболевания бывают затруднения при вставании со стула и ходьбе вверх. Движения, контролируемые дистальными мышцами, страдают позже. Иногда, чаще у детей, возможны внемышечные проявления болезни в виде изъязвлений в желудочно-кишечном тракте и обызвествлений мягких тканей. При полимиозите, так же как при дерматомиозите, поражаются симметричные проксимальные мышцы. Однако при полимиозите нет кожных проявлений. Он встречается главным образом у взрослых. Этиология и патогенез. Этиология воспалительных миопатии неизвестна, но повреждение тканей, видимо, обусловлено иммунными механизмами. При дерматомиозите основной мишенью служат капилляры. Микроциркуляторное русло атакуют антитела и компоненты комплемента, вызывая появление фокусов некроза миоцитов. При полимиозите, наоборот, возникают повреждения, опосредованные клетками. Около поврежденных мышечных волокон найдены CD8+ цитотоксические Т-лимфоциты и макрофаги, а экспрессия HLA-антигенов I класса увеличена на сарколемме нормальных мышечных волокон. Как и при других аутоиммунных заболеваниях, при воспалительных миопатиях выявляются антитела. Диагностика миозита основана на клинических симптомах, Данных электромиографии и биопсии. Смешанные заболевания соединительной ткани. Описаны у тех больных, у которых сочетаются симптомы СКВ, полимиозита и прогрессирующего системного склероза, а серологически наблюдается высокий титр антител к рибонуклеопротеидам. При этих заболеваниях страдают почки; эффективно лечение кортикостероидами. Для смешанных заболеваний соединительной ткани характерны артрит, опухание рук, феномен Рейно, аномальная подвижность пищевода, миозит, лейкопения и анемия, лихорадка, лимфаденопатия и гипергаммаглобулинемия. СИНДРОМЫ ИММУННОГО ДЕФИЦИТА • Иммунодефицитные заболевания — состояния, обусловленные выпадением одного или нескольких компонентов иммунитета. Синдром иммунного дефицита по сути представляет собой эксперимент природы, который еще раз убеждает в сложности устройства иммунной системы. Все иммунодефициты делят на первичные, которые почти всегда детерминированы генетически, и вторичные, связанные с осложнениями инфекционных заболеваний, нарушенным всасыванием, старением, побочными эффектами иммуносупрессии, облучением, химиотерапией и других аутоиммунных болезней. Первичные иммунодефициты. Это генетически детерминированные заболевания. Они поражают специфический иммунитет (гуморальный и клеточный) или неспецифические механизмы защиты хозяина, обусловленные комплементом и клетками (фагоцитами или естественными киллерами). Хотя большинство иммунодефицитов встречается довольно редко, некоторые из них, например дефицит IgA, довольно-таки распространены, особенно у детей. Обычно первичные иммунодефициты проявляются в детстве в возрасте между 6 мес и 2 годами повышенной чувствительностью и рецидивирующими инфекционными заболеваниями. Агаммаглобулинемия Брутона, сцепленная с Х-хромосомой. Является одним из самых распространенных первичных иммунодефицитов и характеризуется фактическим отсутствием сывороточных иммуноглобулинов, хотя IgG могут быть обнаружены, в незначительном количестве. Это заболевание связано с Х-хромосомой и встречается у лиц мужского пола. Тяжелые рецидивирующие инфекции начинаются обычно в возрасте 8—9 мес, когда ребенок перестает получать материнские иммуноглобулины. Чаще всего представлены пиогенные микроорганизмы (стафилококки, Haemophilus influenzae). Больные страдают рецидивирующими конъюнктивитом, фарингитом, средним отитом, бронхитом, пневмонией и кожными инфекциями. С большинством вирусных и грибковых инфекций организм больного справляется успешно, так как клеточный иммунитет не нарушен. Вместе с тем существует особый риск развития связанного с вакцинацией полиомиелита и эховирусного энцефалита, а также пневмоцистной пневмонии. Персистирующая лямблиозная инфекция приводит к нарушению всасывания. При болезни Брутона чаще развиваются аутоиммунные болезни. У половины детей встречаются заболевание типа ревматоидного артрита, а также системная красная волчанка, дерматомиозит и другая аутоиммунная патология. В костном мозге находят нормальное содержание пре-В-лимфоцитов, представляющих собой крупные лимфоидные клетки с IgM в цитоплазме, но без иммуноглобулинов на поверхности клетки; фактически отсутствуют В-лимфоциты, за исключением редких случаев. Лимфатические узлы и селезенка не имеют герминативных центров, а в лимфатических узлах, селезенке, костном мозге и соединительной ткани отсутствуют плазматические клетки. Небные миндалины особенно плохо развиты или рудиментарны. В то же время количество циркулирующих и тканевых Т-лимфоцитов, функция которых не изменена, остается в норме. Общий вариабельный иммунодефицит. Представляет собой гетерогенную группу заболеваний. Он может быть врожденным или приобретенным, спорадическим или семейным (с непостоянным типом наследования). Для всех пациентов характерна гипогаммаглобулинемия, обычно связанная с дефектом всех классов антител, но иногда только IgG. Причины иммунодефицита могут быть различными. В противоположность агаммаглобулинемии Брутона у большинства больных содержание В-лимфоцитов в крови и лимфоидной ткани нормальное. Однако эти В-клетки не могут дифференцироваться в плазматические клетки. В большинстве случаев дефект состоит в терминальной дифференцировке В-лимфоцитов, в результате чего они не могут секретировать нормальное количество иммуноглобулинов даже тогда, когда имеются хелперные Т-лимфоциты, а потенциальные супрессорные Т-лимфоциты отсутствуют. Молекулярная основа аномальной дифференцировки В-лимфоцитов может быть различной. У некоторых больных возникают мутации, которые влияют на экспрессию иммуноглобулиновых генов, у других — дефектные В-лимфоциты, так же как и Функциональные аномалии СD4+-лимфоцитов (хелперов) или СD8+Т-лимфоцитов (супрессоров), причем количество CD4+T-лимфоцитов может быть нормальным, но они продуцируют сниженное количество ИЛ-2 и 7-интерферона (ИФН-γ)- В связи с тем что цитокины необходимы для секреции иммуноглобулинов, указанные дефекты Т-лимфоцитов приводят к гипогаммаглобулинемии. У других больных речь идет не об отсутствии Т-лимфоцитов, а скорее об абсолютном увеличении количества CD8+T-лимфоцитов, которые могут подавлять секрецию антител нормальными В-лимфоцитами. Получены данные о генетической предрасположенности к общему вариабельному иммунодефициту. Клинически заболевание проявляется рецидивирующими инфекциями. Помимо бактериальных инфекций, эти больные страдают тяжелыми энтеровирусными инфекциями, рецидивирующим герпесом и персистирующей диареей, вызванной лямблиями. Гистологически наблюдается гиперплазия В-клеточных участков лимфоидной ткани (лимфоидных фолликулов в лимфатических узлах, селезенке и кишечнике). Расширение этих зон отражает, видимо, дефектную иммунорегуляцию: В-лимфоциты пролиферируют в ответ на антиген, но вследствие нарушенной продукции антител торможение пролиферации посредством IgG отсутствует. У этих больных высока частота аутоиммунных заболеваний, включая ревматоидный артрит, пернициозную и гемолитическую анемию, и составляет примерно 20 %. Изолированный дефицит IgA. Широко распространен. Для заболевания характерен очень низкий уровень как сывороточного, так и секреторного IgA. Иммунодефицит может быть семейным или приобретенным после токсоплазмоза, кори либо некоторых других вирусных инфекций. В связи с тем что IgA является основным иммуноглобулином внешней секреции, при его дефиците нарушается защита слизистых оболочек и развиваются инфекции дыхательной, желудочно-кишечной и мочеполовой систем. Больные нередко страдают синопульмональными инфекциями и диареей. У пациентов с дефицитом IgA аллергия респираторного тракта и различные аутоиммунные болезни, особенно системная красная волчанка и ревматоидный артрит, встречаются очень часто. Причина повышенной частоты аутоиммунных и аллергических заболеваний неизвестна. Основной причиной этого иммунодефицита является дефект дифференцировки В-лимфоцитов, продуцирующих IgA. У большинства больных с селективным дефицитом IgA количество IgA-положительных В-лимфоцитов нормальное, но большинство из них экспрессируют незрелый фенотип, который характеризуется коэкспрессией поверхностных IgD и IgM. Лишь немногие из этих клеток способны in vitro трансформироваться в IgA-плазматические клетки. Сывороточные антитела к IgA обнаружены приблизительно у 40 % больных, что необходимо учитывать при переливании крови, так как при попадании в организм больного крови, содержащей нормальное количество IgA, у него может развиться тяжелая, даже фатальная, анафилактическая реакция. Синдром Ди Джорджи (гипоплазия вилочковой железы). Это пример селективного Т-лимфоцитарного дефицита, появление которого связано с нарушением развития 3-го и 4-го глоточных карманов, дающих начало вилочковой железе, околощитовидным железам, некоторым светлым клеткам щитовидной железы к ультимобранхиальному телу. Таким образом, у этих больных отсутствует клеточный иммунный ответ (вследствие гипоплазии йли отсутствия вилочковой железы), развиваются тетания (отсутствие околощитовидных желез) и врожденные дефекты сердца и крупных сосудов. Кроме того, внешний вид рта, ушей и лица может быть изменен. При отсутствии клеточного иммунитета уровень циркулирующих Т-лимфоцитов низкий и защита против некоторых грибковых и вирусных инфекций слабая. Количество плазматических клеток в лимфоидной ткани нормальное, но тимусзависимые паракортикальные зоны лимфатических узлов и периартериолярных оболочек в селезенке отсутствуют. Содержание иммуноглобулинов в норме. Синдром Ди Джорджи не относится к числу генетически детерминированных заболеваний, но, по-видимому, является результатом внутриматочного повреждения плода на 8-й неделе беременности. Тяжелые комбинированные иммунодефицитные заболевания. Характеризуются комбинированным В- и Т-лимфоцитарным дефектом. Больные дети страдают от тяжелых рецидивирующих инфекций, вызываемые Candida albicans, Pneumocystis carinii, Pseudomonas, а также цитомегаловирусом, вирусом ветряной оспы и многими бактериями. Без пересадки костного мозга смерть наступает в первые годы жизни. В зависимости от локализации мутантного гена и природы генетического дефекта различают два типа наследования: аутосомно-рецессивный и рецессивный, связанный с Х-хромосомой. Приблизительно у 40 % пациентов с аутосомно-рецессивными формами заболевания отсутствует фермент аденозиндеаминаза, дефицит которого ведет к накоплению деоксиаминазина и его производных, которые особенно токсичны для незрелых лимфоцитов, в первую очередь Т-лимфоцитов. Следовательно, количество Т-лимфоцитов может быть заметно снижено в тяжелых случаях. Реже при аутосомно-рецессивном типе этого заболевания встречается дефект активации Т-лимфоцитов. У этих больных содержание Т-клеток нормальное, однако существует дефицит одного из видов молекул, которые участвуют в активации Т-лимфоцитов. Рецессивный тип наследования, связанный с Х-хромосомой, встречается приблизительно у 50 % больных. У них происходит мутация, которая воздействует на белок, являющийся рецепторами для ИЛ-2, ИЛ-4 и ИЛ-7. Характер морфологических изменений зависит от вида генетического дефекта. При двух наиболее распространенных формах иммунодефицита (отсутствие аденозиндеаминазы и мутация рецепторов) вилочковая железа маленькая, лишена лимфоидных клеток. В других случаях лимфоидная ткань гипопластична с заметным уменьшением размеров зон Т-клеток, а в некоторых случаях — как Т-, так и В-зон. Иммунодефицит с тромбоцитопенией и экземой (синдром Вискотта — Олдрича). Это рецессивное, связанное с Х-хромосомой заболевание, которое характеризуется тромбоцитопенией, экземой, уязвимостью к рецидивирующей инфекции и рано заканчивается смертью. Вилочковая железа морфологически нормальна, однако наблюдается прогрессирующее вторичное истощение Т-лимфоцитов в периферической крови и паракортикальных (тимусзависимых) зонах лимфатических узлов с вариабельным снижением клеточного иммунитета. Ответы на такие белковые антигены, как столбнячный и дифтерийный токсин, могут быть нормальными, однако классически они свидетельствуют о слабом антигенном ответе на полисахаридные антигены. Уровень IgM в сыворотке низкий, a IgG — обычно нормальный. Парадоксально возрастает уровень IgA и IgE. У больных часто развиваются злокачественные лимфомы. Генетический дефицит системы комплемента. Описан для всех компонентов данной системы и двух ее ингибиторов. Дефицит компонентов комплемента, особенно СЗ, который необходим как для классического, так и альтернативного пути, обусловливает повышенную чувствительность к инфекции, вызываемой патогенными бактериями. Врожденный дефицит Clq, С2 и С4 повышает риск развития иммунокомплексных заболеваний, например системной красной волчанки. При отсутствии ингибитора С1-эстеразы возникает неконтролируемая активация С1-эстера-зы с образованием кинина С2. У этих больных развивается врожденный ангионевротический отек, характеризующийся поражением кожи и слизистых оболочек. Дефицит компонентов классического пути (С5—8) способствует развитию рецидивирующих нейссеровских (гонококковые, менингококковые) инфекций. Лекция 19
|
||
|
|
Последнее изменение этой страницы: 2017-02-19; просмотров: 248; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.21.46.13 (0.008 с.) |