
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Розділ 1. Особливості поверхні твердого тілаСтр 1 из 35Следующая ⇒
Розділ 1. ОСОБЛИВОСТІ ПОВЕРХНІ ТВЕРДОГО ТІЛА
Склад і будова поверхні
Поверхня реального кристала неоднорідна і характеризується складним мікрорельєфом. На рис.1.1 показані різні деталі рельєфу поверхні. Розглянемо модель, що ілюструє характер положення атомів на поверхні кубічної грати. На такій поверхні атоми можуть знаходитися в наступних положеннях: на повністю заповненому краї - 1; у вершині кута - 2; на цілком заповненій поверхні - 3; на не заповненому краї - 4; у вершині незаповненого кута - 5; на сходинці - 6; на поверхні - 7; вакансії, що виходять на поверхню кристала - 8. Зі всіх положень атома найцікавіше і важливе для практики напівкристалічне положення ½, яке найбільш часто зустрічається при поступовому вирощуванні кристалів і вакуумному нанесення покриттів. В різних процесах різноманітні розташування відіграють різну роль. Наприклад, розміщення ½ дуже важливе для створення вакансій, розміщення 8 суттєве при різних явищах адсорбції, розміщення 3 – типове розміщення на ідеальній поверхні і т. д.
Рис.1.1 Схема можливих характерних положень атомів і вихід дефектів на поверхню кристала з кубічною граткою (модель Косселя і Странського)
Складний рельєф поверхні кристала призводить до того, що на поверхні є атоми, які відрізняються різними числами сусідів та енергією зв’язку (табл. 1.1). Сили зв'язку, які діють на атоми з боку сусідніх атомів, дуже залежать від його положення на поверхні кристала. Повна енергія зв'язку для атома або іона, наприклад, для кубічних кристалічних грат Е =6 Е 1+12 Е 2, Е 1 – енергія зв'язку по поверхні між кубами; Е 2 – енергія зв'язку по гранях куба. Атоми, що знаходяться на поверхні мають меншу енергію зв'язку і залежно від положення атомів її величина зменшується в наступному порядку: Е (3)> E (1)> E (1/2)> E (2)> E (6)> E (7)> E (4)> E (5)
З схеми поверхні кристала можна підрахувати величину енергії атомів в різних положеннях: E (1)=4 E 1+5 E 2; E (2)=3 E 1+3 E 2; E (3)=5 E 1+8 E 2; E (4)= E 1+3 E 2; E (5)= E 1+2 E 2; E (6)=2 E 1+6 E 2; E (7)= E 1+4 E 2 Реальна металева поверхня додатково має велике число дефектів інших типів. До них в першу чергу відносяться дефекти, що виникають в місцях виходу дислокацій і слідів меж зерен та вакансій.
Розділ 3. ЗАГАЛЬНІ ПИТАННЯ ТЕРМОДИНАМІКИ ПОВЕРХНЕВИХ ЯВИЩ Розділ 4. ЯВИЩА АДСОРБЦІЇ
Фізична адсорбція
Сорбційні сили та процеси. Сорбція — це процес поглинання газів або пари твердими чи рідинними тілами. Природа сорбційних сил така ж, як і сил міжмолекулярної взаємодії. Адсорбція відбувається під дією некомпенсованої або міжатомної взаємодії в поверхневому шарі адсорбенту, який притягує молекули адсорбата із приповерхневої області. Адсорбція зменшує поверхневу енергію адсорбента і є початковою стадією процесу взаємодії між твердими речовинами та газом. Розрізняють хімічну і фізичну адсорбції (взаємодію). Фізична взаємодія визначається наступними ефектами: 1. Притягання між молекулою із постійним диполем та молекулою з індуційованим диполем (індукційний ефект); 2. Притягання між молекулами із постійними диполями (орієнтаційний ефект); 3. Притягання між молекулами із флюктуіруючим та індуцірованим диполем (дисперсійний ефект); 4. Відштовхування між ядрами молекул, що зближуються. При хімічній взаємодії розрізняють слідуючи види зв’язків, які забезпечують притягання молекул: ковалентну, металічну, іону. Рідина, яка поглинає газ, називається сорбентом (адсорбентом, абсорбентом). Рідина, що поглинається — сорбат (адсорбат, абсорбат). У випадку фізичної адсорбції між частинкою і поверхнею діють слабкі дальньодіючі сили; енергія зв’язку мала: приблизно 0,1...0,5 еВ.
Основні закони капілярності
Сила поверхневого натягу в простих випадках в залежності від різниці тиску по обидві сторони поверхні з кривизною
де R1 і R2 – головні радіуси кривизни поверхні в даній точці. За допомогою цієї фундаментальної формули можна знаходити надмірний тиск для різних випадків.
Змочування і розтікання Змочування – є особливим видом взаємодії рідини з твердим тілом. Воно відбувається раніше розчинення і дифузії. Виявляється на практиці або у формі розтікання краплі рідини по поверхні твердого тіла, або навпаки у формі відтоку (згортання). Нехай крапля рідини поміщена на ідеально гладку однорідну тверду поверхню, крапля має форму тіла обертання. На рис.5.8 точка А є точка торкання тіла обертання. Дотична АВ до поверхні рідини утворює з поверхнею кут θ, який називається крайовим кутом. Крайовий кут завжди відлічується від дотичної убік рідини. Вершина крайового кута знаходиться на лінії змочування – лінії, уздовж якої стикаються всі фази, що беруть участь в змочуванні (тверде тіло – рідина – газ).
Необхідно розрізняти рівноважні і нерівноважні крайові кути. Рівноважний крайовий кут залежить тільки від термодинамічних властивостей системи, а саме від поверхневого натягу на межах розділу фаз, що беруть участь в змочуванні. Крайові кути, які виміряні при відхиленні системи від стану термодинамічної рівноваги називаються нерівноважними.
Рис. 5.8. Розтікання краплі по поверхні твердого тіла: 1 – початковий (нерівноважний) стан; 2 –рівноважний стан.
Величина рівноважного крайового кута визначається співвідношенням сил притягання рідини до твердого тіла і сил взаємного притягання між частинками самої рідини. Рівноважний крайовий кут представляє одну з найважливіших характеристик змочування. Основними енергетичними характеристиками поверхні твердих тіл і рідин служать питома вільна поверхнева енергія ƒ і поверхневий натяг σ. Питома вільна поверхнева енергія поверхні розділу двох фаз α і β рівна
Різниця Поверхневий натяг σ визначається як робота зворотного ізотермічного процесу утворення одиниці площі поверхні розділу фаз, за умови, що термодинамічні параметри стану системи (Т і Р, хімічні потенціали
де V – об`єм багатокомпонентної системи; nі – число молів і -го компоненту. Питома вільна поверхнева енергія fαβ і поверхневий натяг
де Для однокомпонентних систем
і при Г = 0 По Гіббсу σ, що входить в рівняння, є поверхневий натяг і воно пов'язано з надмірною вільною поверхневою енергією ƒ виразом.
де Гi, µi – адсорбція і хімічний потенціал i -го (компоненту за умови, що поверхня, яка розділяє вибрана так, що адсорбція першого компоненту рівна нулю. Величина σ визначає роботу утворення поверхні, але не її розтягування. Лише для рідини процеси утворення нової поверхні і розтягування еквівалентні. Тому величина σ, хоча і називається поверхневим натягом, для твердого тіла не є механічним натягом (напругою) його поверхні. Останнє через малу рухливість атомів твердого тіла може приводити, наприклад, до зміни міжатомних відстаней в поверхневому шарі, зміни густини поверхневих частинок тіла. Таким чином, величина поверхневого натягу σ, що входить в рівняння, є робота зворотного утворення одиниці площі даної поверхні.
Когезія Часто роботу адгезії зіставляють з роботою когезії Wк. Когезія – притягання між частинками одного і того ж твердого тіла або рідини, обумовлене силами міжмолекулярної взаємодії, що приводить до об'єднання частинок в єдине тіло. Таким чином Wк – характеризує взаємодію частинок однієї фази, тобто визначається силами зв'язку усередині тіла. При ізотермічному розділенні об'єму рідини на дві частини робота розділення з розрахунку на одиницю поверхні рівна 2
Підставивши значення роботи адгезії і когезії в рівняння рівноважного крайового кута, одержимо
Останнє рівняння показує, що величина рівноважного крайового кута визначається співвідношенням сил притягання рідини до поверхні твердого тіла і сил взаємного притягання частинок рідини. Відносна робота адгезії
Основні випадки взаємодії рідини з твердим тілом реалізуються при наступних співвідношеннях робіт адгезії і когезії 1. незмочування 2. змочування 3. повне змочування Робота адгезії завжди позитивна, оскільки між тілами будь-якої природи завжди діють сили молекулярного притягання.
Зародкоутворення Основною перепоною на шляху фазових перетворень є необхідність утворення центрів нової фази чи зародку нової фази. Буває гомогенне і гетерогенне зародження. Гомогенна система – термодинамічна система, всередині якої нема поверхонь розділу, відділяють частини системи друг від друга. Гетерогенна система це неоднорідна термодинамічна система, яка складається з однорідних фаз, розділених поверхнею розділу. Основною перешкодою на шляху фазових перетворювань являється необхідність утворення центрів нової фази або зародку нової фази. Центри чи зародки нової фази утворюються на макроскопічній поверхні основи, і їх утворення в багато чому залежить від поля сил цієї поверхні, і їх зародження являється гетерогенним. Зародкоутворення починається з того, що на поверхні виникає скупчення атомів, котрі затримуються в потенціальних ямах, розташовуючись на відстанях, кратних міжатомним відстаням основи. Між атомами в скупченні виникають сили хімічного зв’язку, які намагаються зблизити ці атоми. Однак взаємодія адсорбованих атомів поверхні основи призведе до появи сил, направлених в протилежний напрямок і які намагаються розтягнути скупчення. Джерелом цих сил є те, що мінімум вільної енергії поверхні розділу «скупчення – основа» спостерігається тоді, коли адсорбовані атоми точно розташуються в потенціальних ямах і взаємодія між ними мінімальна. Тому поява скупчень атомів призводить до збільшення вільної енергії системи. Виникаючі сили поверхневого натягу на межі «скупчення—пар» і «скупчення—основа» намагаються стягнути скупчення в компактне утворення – зародок. Їм протидіють сили поверхневого натягу основи.
Припущення, що процес кристалізації полягає в утворенні центрів (зародків) кристалізації і в їх зростанні, вперше висказав більше 100 років тому Д. К. Чернов. Перш ніж розглянути, як це проходить, необхідно розібрати, в чому принципіальна різниця в побудові рідкої і кристалічної фаз. Встановлено, що ця різниця полягає в ступіні порядку в розташуванні частинок. Розміщення атомів в кристалі і в рідині відносно якогось конкретного атома приведено на рис.6.4 (а,б). У випадку кристалічного вкладення атомів (рис.6.4,а) існує порядок в їх розташуванні, як поблизу атома А, так і здалеку від нього. Тому кажуть про наявність в кристалі, як ближнього, так і дальнього порядку упаковки. Для рідкої фази (рис.6.4,б) поблизу атома А також спостерігається порядок (шість атомів своїми центрами розташовуються близько від кола, проведеного із центра атома А). З віддаленням від атома А порядок все більше порушується. Тому можна говорити про наявність у рідини ближнього і відсутність дальнього порядку.
Як відмічалось, процес кристалізації складається з утворення центрів кристалізації і росту цих центрів, що залежить від ступеня переохолодження Δ T. На рис. 6.5 показана залежність числа центрів (Ч.Ц) і швидкості росту кристалів (Ш.Р) від ступеня переохолодження Δ T. Із рисунка видно, що центри кристалізації утворюються тільки при температурі нижче теоретичній - Ts. Співставляючи рис.6.2 та 6.5, можна зробити висновок, що при T>TS атоми перебудовуються, створюючи в окремих ділянках кристалічний стан, тобто існує ближній і дальній порядки. Термодинаміка (рис.6.2) показує необхідність цього, так як G т< G р, але не пояснює механізму процесу, утворення центрів кристалізації. Як при T>TS, так і при T<TS атоми рухаються. При T>TS більшість атомів групується в ближньому порядку, але це не значить, що в окремих місцях не виникає також дальній порядок. Таким чином, керуючись категоріями “закономірність” і “випадковість”, можна сиверджувати, що і в рідкій фазі при T>TS існують утворення, подібні кристалічним. При G P< G T ці утворення нестійкі, розпадаються в одному місці і виникають в інших ділянках. При T > Ts кристалічні утворення становляться стійкими і будуть зростати, якщо їх ріст буде супроводжуватись зменшенням термодинамічного потенціалу.
Щоб судити про це, потрібно проаналізувати баланс потенціалу G при утворенні кристалу. Якщо загальна зміна ізобарного потенціалу Δ G при кристалізації складається із двох складових Δ GV і Δ GS, то Δ G = GV +Δ GS, де Δ Gv – зменшення потенціалу за рахунок утворення твердої фази об’ємом V; тобто Δ GV <0; Δ GS – приріст потенціалу із-за утворення поверхні розділу між твердою і рідкою фазами, в котрій діє поверхневий натяг σ, Δ GS >0. Якщо загальна поверхня розділу фаз F, то G S = σ F.
Рис. 6.5. Вплив ступеня переохолодження Δ T на число центрів кристалізації (Ч. Ц.) і швидкість росту кристалів (Ш. Р.).
Із приведених умов можна скласти рівняння енергетичного балансу системи, що кристалізується Δ G=–V(G P– G T)+σ F (6.5)
Із рис.6.6 видна залежність ізобарного потенціалу G від розміру кристалу при різних значеннях Δ T. Кожній температурі відповідає критичний розмір зародку, зростання якого термодинамічно виправдано. З цієї залежності витікає, що процес кристалізації повинен протикати тим інтенсивніше, чим більша ступінь переохолодження (>Δ G) і менше коефіцієнт поверхневого натягу σ. На рис.6.5 приведена крива залежності числа центрів кристалізації (Ч.Ц) від ступеню переохолодження Δ T. Штрихова частина кривої Ч.Ц. при кристалізації металів в звичайних умовах не реалізується. Розглянемо тепер іншу частину механізму процесу кристалізації — ріст кристалів. Ріст кристалів виконується утворенням на гранях кристалу двомірних зародків. Мінімальний розмір їх зменшується зі збільшенням степені переохолодження. Крива (Ш.Р.) на рис.6.5 має таку ж форму, як і крива Ч.Ц. Тільки максимум кривої Ш.Р. зсунутий до більш високих значень степеня переохолодження Δ Т. Для металів реалізується частина кривої Ш.Р., що йде вгору. Багато неметалічних матеріалів з крупними частинками, молекулами (скло, полімери, смоли) схильні до дуже великих переохолоджень. Вони поступово знижають рідинотекучість, твердіють, не набувають кристалічної будови. Такі тіла називаються аморфними. Метали можливо також отримати в аморфному стані, але для цього потрібні дуже високі степені переохолодження.
Критичний розмір зародку. Гомогенне зародкоутворення. Частинка нової фази, яка виникає із скупчення, для котрої робота утворення максимальна, є критичним зародком. Умова максимуму роботи співпадає з умовою рівноваги зародку. Рівновага ця нестійка, так як збільшення розмірів зародку призводить до його наступного росту, а зменшення — до розпаду. Допускається, що утворені зародки мають рівноважну форму – куполоподібну. Розглянемо зміну вільної енергії Δ F при утворенні гомогенного зародку (котрий не взаємодіє з поверхнею). Розглядається переохолоджена фаза, яка повинна перетворитися в нову фазу, що має приблизно таку ж густину, але меншу вільну енергію на одиницю об’єму, тобто Δ FV негативно. Якщо ця нова фаза являє собою сферу радіуса r, то зміна вільної енергії складе Таким чином, повну зміну вільної енергії Δ F, що виражена як функція радіусу, можна уявити складеної з двох складових — зміна поверхневої і об’ємної енергії у виді:
Радіус r в рівнянні (6.6) за умови, що функція
Із (6.6) отримаємо
Критичний радіус
При цьому значення вільної енергії має вигляд
Функція Охолодження до температури, меншої температури рівноваги, тобто переохолодження, робить величину
Рис.6.6. Критичний радіус зародку нової фази (гомогенне зародження) Збільшення зародку нової фази до критичного радіусу
В той же час надто низька температура настільки затрудняє рух атомів, що швидкість утворення зародків нової фази знову зменшується. З цього випливає, що крива залежності швидкості утворення зародків нової фази від температури переохолодження повинна мати максимум.
Рис. 6.7. Вплив переохолодження на величину критичного радіусу.
Виходячи з рис.6.7, видно, що критичний радіус
На утворення зародків при конденсації на поверхні основи більше впливає тип взаємодії між матеріалом основи і зародком, що проявляється через рівноважний кут q. При хімічній взаємодії відбувається змочування і зародок добре розтікається по поверхні. В цьому випадку Ð q ® 0. При цьому D F кр ® 0. З цього випливає, що утворення зародку буде полегшене. Якщо змочування не проходить, Ðq ® 180°, а D F збільшується і утворення зародку буде ускладнено. Чистота поверхні сильно впливає на контактний Ð q, впливаючи на умови конденсації і утворення зародків. В загальному випадку ріст критичного зародку може виникати: 1) шляхом прямого захвату атома з пари. 2) шляхом приєднання дифундуючих по поверхні адсорбованих атомів; останній процес домінує при збільшенні температури. Загальні положення Внаслідок флуктуації теплової енергії деякі атоми у твердому тілі можуть отримувати енергію більшу середнього значення, яке мають атоми у вузлах кристалічної гратки. Такі атоми здатні переборювати потенційні бар'єри, що підтримують їх у рівноважних положеннях у кристалі. Залишивши свої місця атоми займають міжвузлове положення, а вакансію, яка утвориться при цьому, займає один із сусідніх атомів, що, очевидно, обумовлює переміщення вакансії в кристалі тобто відбувається перенос речовини. Таким способом атоми у твердому тілі можуть переміщатися – дифундувати, зі швидкістю, що збільшується з температурою. Дифузія – розповсюдження речовини в якому не будь середовищі в напрямку убування його концентрації, обумовлене тепловим рухом атомів, молекул, іонів і інших більш великих часток. Дифузія можлива після змочування і розтікання рідкої фази по поверхні твердого тіла. Переміщення атомів в наслідок теплових коливань атомів, перескоки їх у сусідні позиції з частотою теплових флуктуацій зберігаються при всіх видах дифузії. Елементарний акт дифузії складається в стрибкоподібному переміщенні атома на відстань, порівнянній з періодом ґрат. Дифузійні процеси класифікують декількома способами. Розрізняють самодифузію і гетеродифузію. Ефект самодифузії має місце в однокомпонентних системах і складається з флуктуаційних перескоків однотипних атомів, що відбуваються й у відсутності градієнта концентрації. Гетеродифузія – ефект переміщення хімічно різнорідних атомів. На рис.7.1. зображено розподіл дифундуючих компонентів в контакті двох металів – міді і нікелю.
а) б) в)
Рис.7.1. Різні стадії дифузії в системі Cu – Ni: а – вихідний стан; б – нагрівання при 1273 К; в – кінцевий стан.
Розрізняють зовнішню і внутрішню дифузію. Внутрішня дифузія – переміщення атомів в нутрі відповідної кристалічної фази. Зовнішня дифузія – флуктуаційний перехід атомів із однієї фази в другу і відбувається на границі цих фаз. Відома дифузія по поверхні, що також здійснюється флуктуаційним шляхом, але при цьому необхідна теплова флуктуація тут менше, тому що робота, необхідна для того, щоб відірвати атом від його сусідів при перескоку в наступну позицію, на поверхні менша. З іншого боку, кількість атомів, що беруть участь у поверхневій дифузії, менша, ніж у випадку об'ємної дифузії. Вздовж границі двох різноорієнтованих кристалів однієї і тієї ж фази швидкість дифузійного переміщення атомів також буде відрізнятися від швидкості внутрішньої дифузії в зв'язку зі зміненим значенням теплової флуктуації, необхідної для перескоку атомів, і зміненим значенням числа атомів, що беруть участь у дифузії. Подібну дифузію називають граничною. Якщо мається границя двох кристалів, що належать до різних фаз, то вздовж неї може мати місце відмінна по швидкості від інших видів дифузії гранична міжфазна дифузія. Закони дифузії Фіка Поступальна дифузія. Перший закон Фіка. Поступальна дифузія – це самовільний процес. Експериментально встановлено, що, як тільки в середовищі виникає неоднорідність по концентрації, починається спонтанна дифузія, що прагне усунути цю неоднорідність. Дифузія протікає відповідно до закону Фіка. Дифузія спостерігається при нерівномірному розподілі компонентів, що дифундують, у матеріалі – в результаті може вийти спрямований потік атомів через наявність градієнта концентрацій або хімічного потенціалу. Цей процес відбувається на границі двох фаз і складається з переходу через границю фаз (рис.7.2, в).
Рис.7.2. Модель для описання розповсюдження речовини вздовж осі Х, згідно зпершим і другим законами Фіка: а – залежність концентрації; б – залежність потоку речовини, що відповідає концентрації; в – елемент об’єму, в який входить потік J1 і виходить J2.
Потік через задану площину пропорційний градієнту концентрації. Якщо градієнт концентрації першого компонента спрямований по осі х, то потік цього компонента уздовж цієї ж осі (J) дорівнює:
де D – коефіцієнт дифузії. Це рівняння називають першим законом Фіка. Члени даного рівняння мають наступну розмірність Другий закон Фіка Якщо стаціонарний стан не досягнутий, тобто концентрація в кожній точці міняється в залежності від часу, то більш зручно використовувати інше диференціальне рівняння, що виходить з рівняння балансу речовини. Розглянемо одиничну площадку, розташовану перпендикулярно осі Х (рис.7.2). Швидкість нагромадження речовини в цій зоні дорівнює швидкості збільшення концентрації в часі C = f (t), помноженої на елементарний об’єм, у котрий входить потік. Нехай J 1 – потік речовини через цю площадку в перетині х, а J 2 – у перетині х +D х. Тоді швидкість нагромадження речовини дорівнює
або різниці потоків
Вираз (7.3) характеризує збільшення кількості речовини в елементі об’єму Використовуючи ці рівняння і перший закон Фіка, одержимо
або
Далі з врахуванням першого закону Фіка одержуємо
Рівняння (7.6) є вираження другого закону Фіка. Тут коефіцієнт дифузії D не залежить від концентрації і має те саме значення в усіх напрямках. Таким чином у деякій точці в даний момент часу швидкість зміни концентрації в часі
Дане вираження відноситься тільки до одномірної дифузії, а коефіцієнт дифузії не залежить від концентрації.
Деякі рішення законів Фіка Якщо величина коефіцієнта дифузії D не залежить від координат, то другий закон Фіка прийме вид
1. У стаціонарному стані
2. Як частне рішення для лінійного розміру R, що характеризує область, зайняту речовиною, що дифундує, одержуємо наступний закон його збільшення зчасом:
Значення коефіцієнта К може змінюватися від 2 до Далі, якщо позначити через М повну масу речовини, що дифундує, то із співвідношення М~ρСR3 і попереднього (7.33)одержуємо закон, по якому концентрація убуває з часом в процесі розповсюдження речовини, що дифундує:
Характерна швидкість дифузії речовини, що займає об’єм порядку R3:
3. Розглянемо застосування закону до дифузійної пари, у якої в одному об’ємі концентрація компонента, що дифундує, в момент часу t=0 дорівнює С0, а в іншому об’ємі - нулеві. Рішення диференціального рівняння (7.32) буде мати вигляд:
де С0 – вихідна концентрація компонента, що дифундує; w- перемінна інтегрування. Вираз
називається функцією помилок Гауса. Значення функції помилок беруться з довідника.
Поверхнева самодифузія В області високих температур потік маси може здійснюватися, як в наслідок переміщення легко рухливих атомів, що знаходяться в стані адсорбції (адатоми), так і внаслідок переміщення атомів у тонкому приповерхньому шарі, де в зв'язку з наявністю дефектів структури дифузійна рухливість атомів перевищує їхню рухливість в об’ємі. Це приповерхня самодифузія (поверхнева самодифузії). Поверхнева дифузія або процес міграції атомів по “гладкій “ поверхні відбувається неупорядковано. У цьому процесі атоми поводяться подібно броунівським часткам. У рівноважних умовах вздовж поверхні відсутній градієнт хімічного потенціалу При наявності градієнта хімічного потенціалу вздовж поверхні (зокрема обумовленого її кривизною), дифузійна міграція стає спрямованої в тому смислі, що кожна з часток, приймаючи участь у хаотичному тепловому блуканні, здобуває деяку швидкість у визначеному напрямку, таким чином, виникає спрямований потік адатомів. Локальне значення хімічного потенціалу атома, розташованого на викривленій ділянці поверхні, залежить від кривизни цієї ділянки і визначається співвідношенням:
де Спрямований дифузійний потік є спрямованим потоком точкових дефектів, що здійснюються під впливом різниці хімічного потенціалу, при цьому перенос маси внаслідок спрямованого потоку адатомів може здійснюватися лише в міру спрямованого переміщення ступіней. Вираз для дифузійного потоку записується у виді (у випадку одномірного руху):
Швидкість переміщення ступіні v зв'язана з коефіцієнтом поверхневої дифузії адатомів
де
Компоненти сплаву у відповідність із їхніми властивостями можуть вступати у хіміко-фізичну взаємодію із середовищем і залишати поверхневі шари тіла, обумовлюючи зміну його властивостей (наприклад, поява зневуглецеваного шару). Групи середовищ Відповідно до відомої класифікації, робочі середовища (рідкі та газові) при заданих умовах поділяються на такі групи. 1. Неактивні середовища. Тобто такі які не впливають на властивості тіла. До них відносяться деякі спектpально чисті інертні гази (аргон, гелій). 2. Поверхнево-активні середовища (фізично активні), тобто такі які впливають на властивості тіла, але не вступають із ними в хімічну взаємодію; Це хімічно неактивні або слабко активні середовища, такі як розчини поверхнево-активних речовин у вуглеводні та у деяких випадках у воді, а також деяких легкоплавких рідких металів. Із цих середовищ виникає адсорбція повеpхнево-активних елементів на поверхні металу, а також у середині його, на стінках дефектів, має місце руйнування металів, адсорбційна втома та ряд інших явищ. 3. Коpозійно-активні (хімічно активні) середовища, тобто, такі які впливають на властивості тіла в результаті хімічної взаємодії з ним (виникнення хімічних зв'язків). До них належать деякі гази, вода та інші водні розчини (електроліти), розплави солей. Вони викликають хімічну або електрохімічну корозію, знижуючи пластичність, міцність і довговічність конструкційних металів. У цих середовищах може спостерігатися розтріскування металу, який перебуває під механічним напруженням (у тому числі під залишковими напруженнями) і корозійна втома металу. Збільшення темпеpатуpи підсилює корозійний вплив на метал. 4. Середовища які адсорбуються об'ємом металу, але не вступають із ним у хімічний зв'язок і не утворюють твердих розчинів. До таких середовищ ставиться водень, який оклюдується сталлю, і що викликає явище водневої крихкості сталі. 5. Середовища, які розчиняють конструкційний метал.
|
||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2017-02-07; просмотров: 189; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.223.20.57 (0.151 с.) |


 виражається формулою Лапласа
виражається формулою Лапласа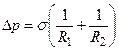 5.1
5.1
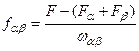 (5.8)
(5.8) – енергія Гельмгольца фази α; F (β) – енергія Гельмгольца фази β; F – загальна енергія Гельмгольца двофазної системи, що має поверхню розділу фаз площею
– енергія Гельмгольца фази α; F (β) – енергія Гельмгольца фази β; F – загальна енергія Гельмгольца двофазної системи, що має поверхню розділу фаз площею  ;
; 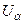 , Tα, Sα – це внутрішня енергія, температура і ентропія фази α.
, Tα, Sα – це внутрішня енергія, температура і ентропія фази α. F – (Fα + Fβ)
F – (Fα + Fβ)  є загальною вільною енергією поверхневого шару Fпш.
є загальною вільною енергією поверхневого шару Fпш.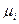 компонентів) не змінюються. Таким чином
компонентів) не змінюються. Таким чином (5.9)
(5.9) зв'язані співвідношенням
зв'язані співвідношенням (5.10)
(5.10) – хімічний потенціал компоненту; Г і – адсорбція цього компоненту на поверхні розділу фаз (надлишок концентрації компоненту i в поверхневому шарі в порівнянні з його концентрацією в об'ємі).
– хімічний потенціал компоненту; Г і – адсорбція цього компоненту на поверхні розділу фаз (надлишок концентрації компоненту i в поверхневому шарі в порівнянні з його концентрацією в об'ємі). (5.11)
(5.11) (5.12)
(5.12) (5.13)
(5.13)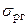 (коефіцієнт 2 враховує, що утворюються дві нові поверхні). Тоді
(коефіцієнт 2 враховує, що утворюються дві нові поверхні). Тоді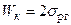 (5.25)
(5.25) (5.26)
(5.26)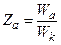 (5.27)
(5.27)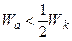
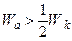
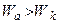 (5.28)
(5.28) а)
а)
 б)
б)

 . Однак, зародок нової фази має поверхню
. Однак, зародок нової фази має поверхню  , і поверхнева енергія буде дорівнювати
, і поверхнева енергія буде дорівнювати  , де
, де  – енергія, на одиницю площі поверхні.
– енергія, на одиницю площі поверхні. (6.6)
(6.6) (6.7)
(6.7)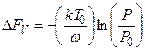 (6.8)
(6.8) = 1 буде дорівнювати критичному
= 1 буде дорівнювати критичному  при
при (6.9)
(6.9) (6.10)
(6.10)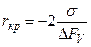 (6.11)
(6.11)
 (6.12)
(6.12) виражається кривою з максимумом. Для найпростішого сферичного зародку початковий період утворення буде супроводжуватись збільшенням вільної енергії, поки він не досягне критичного радіуса
виражається кривою з максимумом. Для найпростішого сферичного зародку початковий період утворення буде супроводжуватись збільшенням вільної енергії, поки він не досягне критичного радіуса  більш від’ємною і, таким чином значно зменшує як критичний радіус, так і енергетичний бар’єр, пов’язаний з утворенням зародку нової фази (рис.6.7). При цьому зростає
більш від’ємною і, таким чином значно зменшує як критичний радіус, так і енергетичний бар’єр, пов’язаний з утворенням зародку нової фази (рис.6.7). При цьому зростає 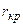 вірогідність придбання необхідним числом атомів потрібної енергії за рахунок яких-небудь локальних флуктуацій. Як наслідок виникає реакція, котра не могла початися при рівноважній температурі, причому ця реакція проходить тим легше, чим більша степінь переохолодження.
вірогідність придбання необхідним числом атомів потрібної енергії за рахунок яких-небудь локальних флуктуацій. Як наслідок виникає реакція, котра не могла початися при рівноважній температурі, причому ця реакція проходить тим легше, чим більша степінь переохолодження.




 (7.1)
(7.1)
 (7.2)
(7.2) (7.3)
(7.3) .
. (7.4)
(7.4) (7.5)
(7.5) (7.6)
(7.6) зв'язана з другою похідною градієнта концентрації через коефіцієнт дифузії D
зв'язана з другою похідною градієнта концентрації через коефіцієнт дифузії D
 (7.32)
(7.32) , і рішення рівняння (7.32) зводиться до одномірної задачі з прямокутними координатами
, і рішення рівняння (7.32) зводиться до одномірної задачі з прямокутними координатами (7.33).
(7.33). (7.34)
(7.34)
 (7.35)
(7.35) (7.36)
(7.36) (7.37)
(7.37) =
=  (7.38)
(7.38) .
.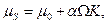 (7.47)
(7.47)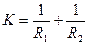 – кривизна поверхні, R1 і R2 – головні радіуси кривизни поверхні;
– кривизна поверхні, R1 і R2 – головні радіуси кривизни поверхні;  – хімічний потенціал атома, розташованого на плоскій ділянці поверхні (R1,2 =
– хімічний потенціал атома, розташованого на плоскій ділянці поверхні (R1,2 = 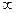 );
); 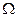 – атомний обсяг;
– атомний обсяг;  – поверхнева енергія.
– поверхнева енергія. (7.48)
(7.48) і градієнтом хімічного потенціалу
і градієнтом хімічного потенціалу 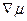 наступним співвідношенням
наступним співвідношенням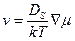 (7.49)
(7.49)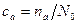 – поверхнева концентрація адатомів, nа і N0 – відповідно число адсорбованих атомів і число атомних позицій на одиниці площі поверхні, S – ділянка дуги в напрямку потоку.
– поверхнева концентрація адатомів, nа і N0 – відповідно число адсорбованих атомів і число атомних позицій на одиниці площі поверхні, S – ділянка дуги в напрямку потоку.


