Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Этиология и патогенез. Для синдрома Гурлер характерен аутосомнорецессивный тип наследования, при синдроме Гунтера — наследование рецессивное, сцепленное с полом.Содержание книги
Поиск на нашем сайте
Установлено, что в основе Гаргоилизма лежат генетически обусловленные нарушения обмена кислых мукополисахаридов, связанные с нарушением структуры, избыточным накоплением их в тканях организма и повышенным выведением с мочой. Определённое значение в патогенезе отводится ферментам, участвующим в расщеплении кислых мукополисахаридов.
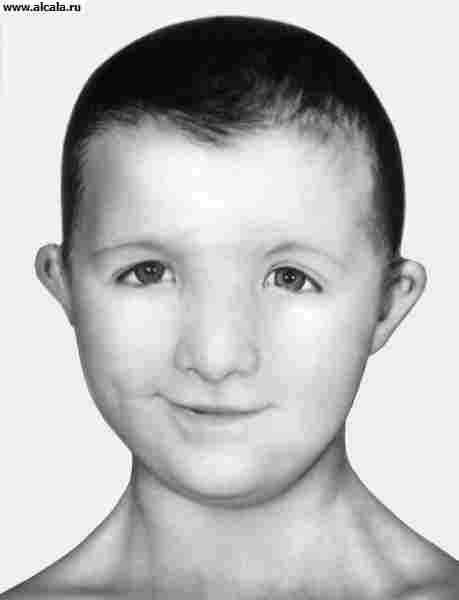
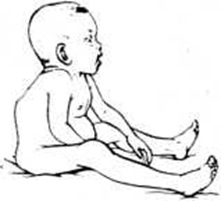
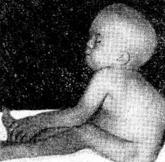
Клиническая картина. При синдроме Гурлер больной имеет характерный внешний вид. Уже на первом году жизни развиваются типичные для данного заболевания изменения конфигурации черепа, деформация позвоночника (см. рисунок). Эти изменения часто расцениваются как признаки рахита. К одному году состояние больных ухудшается, поясничный кифоз и тугоподвижность суставов резко усиливаются. Дети отстают в физ. и умственном развитии. После одного года - симптомы становятся особенно выраженными. Для больных гаргоилизмом характерны: запавшая переносица, нависший лоб, толстые губы и язык, широко расставленные зубы, деформация и увеличение в размерах черепа, отставание в росте, нижнегрудной и поясничный кифоз, деформация грудной клетки, тугоподвижность суставов, пупочные и паховые грыжи, гепато- и спленомегалия, изменения со стороны сердца (систолический шум, приглушение тонов, расширение границ сердца).
Рис. Ребёнок, больной гаргоилизмом (синдром Гурлер); выражена деформация позвоночника.
На ЭКГ выявляется диффузное поражение миокарда (т.е. сердца). Поражение органа зрения наблюдается у всех больных и является наиболее ранним и типичным симптомом заболевания.
Прогноз. Болезнь неуклонно прогрессирует. Летальный исход наступает от заболеваний дыхательных путей и сердечной декомпенсации. Наиболее тяжёлым является первый тип мукополисахаридоза (синдром Гурлер), характеризующийся прогрессирующим течением и ранней смертью больных (большинство погибает до 10—12-летнего возраста). Мукополисахаридоз второго типа (синдром Гунтера) протекает менее злокачественно — больные доживают до 30 и более лет. При проведении ранней терапии прогноз более благоприятный. Профилактика. Родителям, имеющим больного ребёнка, рекомендуется воздержаться от дальнейшего деторождения. Возможна внутриутробная диагностика заболевания на третьем месяце беременности
Наследственные генные болезни Моногенные болезни наследуются в соответствии с законами классической генетики Менделя. Соответственно этому, для них генеалогическое исследование позволяет выявить один из трёх типов наследования: аутосомно-доминантный, аутосомно-рецессивный и сцепленное с полом наследование. Это наиболее широкая группа наследственных заболеваний. В настоящее время описано более 4000 вариантов моногенных наследственных болезней, подавляющее большинство которых встречается довольно редко
При аутосомно-доминантном типе наследования действие мутантного гена проявляется практически всегда. Вероятность развития болезни в потомстве составляет 50%. Больные мальчики и девочки рождаются с одинаковой частотой. Один из родителей больного ребенка обязательно болен. Для аутосомно–доминантного типа наследования характерно: - Проявление признака у гетерозиготных носителей гена. - При анализе родословной признак выявляется в каждом поколении. - Различная выраженность клинических проявлений не только между разными семьями, но и внутри каждой семьи. - Клинические признаки могут появиться не сразу после рождения, а спустя много лет. - Здоровые члены семьи не могут иметь больных детей. пример: Синдром Нунен Синдром Нунен
Лицо ребенка с синдромом Нунен: видны характерные для этого заболевания широкая переносица, антимонголоидный разрез глаз, короткая шея. Заболевание выявляется как у мужчин, так и женщин. Кариотип (хромосомный набор) - 46ХХ или 46XY соответствует полу. Нунен синдром наследственное заболевание, характеризующееся низкорослостью и аномалиями развития. Описан Ж. Нунен в 1963 г. Может быть спорадическим или семейным, наследуемым по аутосомно-рецессивному типу. Измененные гены, локализуются в аутосомах. Заболевание выявляется как у мужчин, так и женщин. Основными клиническими проявлениями являются низкорослость, крыловидные складки кожи на шее, низкая граница роста волос на затылке, птоз, антимонголоидный разрез глаз, деформация или низкая посадка ушных раковин, асимметрия лица, широкая переносица, что делает больных похожими друг на друга. Характерны короткая шея, плоскостопие, изменение формы и структуры позвонков и другие аномалии, отмечаемые при Шерешевского — Тернера синдроме. (Пороки сердца врождённые) У девочек часто наблюдается позднее начало менструаций Задержка умственного развития. Кариотип 46ХХ или 46XV соответствует полу. Диагноз устанавливают на основании характерных клинических признаков и данных лабораторных исследований.
При аутосомно-рецессивном типе наследования мутантный ген проявляется только в гомозиготном состоянии, когда один рецессивный ген ребенок получает от отца, а второй - от матери. Вероятность рождения больного ребенка составляет 25%. Больные мальчики и девочки рождаются с одинаковой частотой. Родители больных детей могут быть внешне здоровы, но являются гетерозиготными носителями мутантного гена. Аутосомно-рецессивный тип наследования наиболее характерен для болезней обмена, при к-рых нарушена функция одного или нескольких ферментов Для аутосомно–рецессивного типа наследования характерно: - Мутантный ген проявляется только у гомозигот по рецессивному гену. - Если родители гетерозиготны, то вероятность рождения больного ребенка составляет 25%. - При анализе родословной мутантный ген проявляется не в каждом поколении. - Вероятность проявления мутантного гена возрастает в родственных браках. - Частота проявления мутантного гена у лиц женского и мужского пола одинакова.
Фенилкетонурия (ФКУ) – наследственное заболевание обмена, характеризующееся поражением ЦНС и прогрессирующим, особенно в первые 2–3 года жизни, слабоумием. Фенотипически здоровые родители больного ребенка являются гетерозиготными носителями мутантного гена. Частота заболевания в Европе в среднем составляет 1: 10 000 новорожденных. ФКУ наблюдается примерно у 1% умственно отсталых лиц.
Заболевание обусловлено мутацией гена, контролирующего синтез фермента фенилаланингидроксилазы, который обеспечивает превращение поступающего в организм с пищей фенилаланина в тирозин
Нарушение последнего процесса приводит к резкому повышению содержания фенилаланина в сыворотке крови и спинномозговой жидкости, при этом отмечают дефицит тирозина, что определяет недостаточный синтез гормона щитовидной железы и меланина, при недостаточном количестве которого наблюдается слабая пигментация кожи и волос. При ФКУ нарушается также синтез серотонина, что губительно действует на нормальное функционирование нервной системы.
Дети с ФКУ рождаются с полноценным головным мозгом, так как биохимические процессы плода осуществляются за счет процессов в организме матери. Возникающие после рождения биохимические нарушения оказывают токсическое воздействие на нервную систему, в результате чего нарушаются процессы миелинизации, развитие и рост мозга.
Нарастание интеллектуального дефекта сочетается с отставанием в физическом развитии, часто с признаками умеренной микроцефалии. Характерен внешний вид больных (блондины со светлой кожей и голубыми глазами) и отдельные диспластические признаки (высокое небо, эпикант, деформация ушных раковин).
При этом отмечают следующие неврологические нарушения: мышечную гипертонию, повышение сухожильных рефлексов, гиперкинезы, тремор пальцев рук, атаксию, нарушения черепно–мозговой иннервации. В более редких случаях имеет место мышечная гипотония; судорожный синдром наблюдается у 20–50% больных.
Уровень интеллектуального развития колеблется от нормы до глубокой идиотии. Динамика слабоумия наиболее выражена в первые 2–3 года жизни. Больные отличаются инертностью, недостаточной целенаправленностью с характерными нарушениями внимания, памяти, недоразвитием гностических функций и пространственных представлений.Отмечается также выраженное недоразвитие речи и нарушения звукопроизношения. Нарушения речи обычно сопоставимы с глубиной интеллектуального дефекта.
|
|||||
|
|
Последнее изменение этой страницы: 2021-05-26; просмотров: 281; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.145.97.1 (0.01 с.) |

 Моногенные болезни, наследуемые по аутосомно–доминантному типу
Моногенные болезни, наследуемые по аутосомно–доминантному типу