Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Алгоритм лабораторного генетичного обстеження пацієнтів з підозрою на ссаСодержание книги
Поиск на нашем сайте
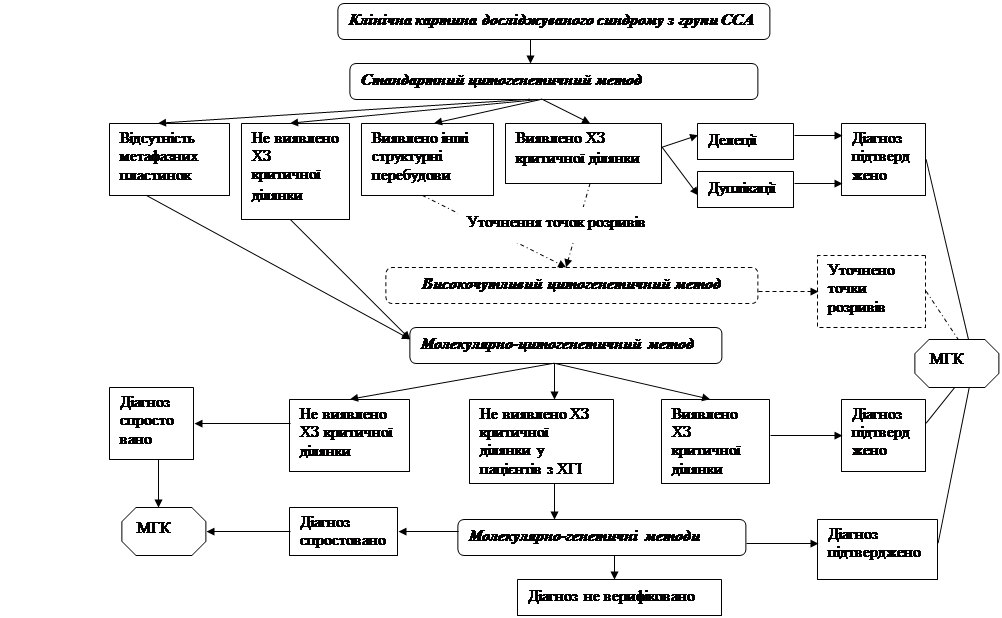
Перший етап лабораторного генетичного обстеження – цитогенетичний аналіз. Усім пацієнтам із попереднім клінічним діагнозом ССА проводять стандартне цитогенетичне дослідження. · У разі виявлення делеції/дуплікації критичних ділянок хромосом 4, 5, 7, 15, 17, 22 або їхньої участі у структурних перебудовах з іншими хромосомами з втратою критичної ділянки діагноз підтверджується. Пацієнт та члени його родини направляються для завершення медико-генетичного консультування до лікаря-генетика. · При виявленні інших структурних перебудов, у яких критична ділянка не задіяна та при необхідності встановлення точок розривів-з’єднання можливим є використання високочутливого цитогенетичного аналізу. · Виявлення структурних перебудов у каріотипі дитини обумовлює необхідність каріотипування батьків дитини, що дозволяє встановити батьківське походження перебудованих хромосом та уточнити точки розривів у хромосомах, які брали участь у структурній перебудові. · При відсутності метафазних пластинок на хромосомних препаратах одразу переходять до другого етапу – використовують FISH-аналіз на інтерфазних ядрах. Варто зазначити, що для пацієнтів, у яких діагноз був підтверджений за допомогою FISH-методу на інтерфазних ядрах, у подальшому ми рекомендуємо провести стандартний цитогенетичний аналіз із метою виключення інших структурних перебудов у каріотипі, оскільки вони можуть впливати на стан дитини. При характерній клінічній картині ССА і нормальному каріотипі переходять до другого етапу – молекулярно-цитогенетичної діагностики. · FISH-діагностику проводять усім пацієнтам з попереднім клінічним діагнозом ССА, у яких на першому етапі не виявлено делецію критичної ділянки досліджуваної хромосоми. · При виявленні мікроделеції критичної ділянки діагноз підтверджується, і родина повертається до лікаря-генетика для подальшої консультації. · Якщо мікроделецію критичної ділянки не виявлено, діагноз спростовується, і пацієнт направляється до лікаря-генетика. У цьому випадку найвірогідніше, що
пацієнт має патологію іншого генезу і тому потребує подальшої диференційної діагностики. · Винятком із цього правила є пацієнти з хворобами геномного імпринтингу (наприклад, синдром Прадера-Віллі). Якщо під час проведення FISH-аналізу не виявлено мікроделеції в каріотипі пробанда з попереднім клінічним діагнозом СПВ, це є приводом для подальшого дослідження із застосуванням методів молекулярного аналізу з метою виявлення функціональної делеції, уніпарентної дисомії або мутації центру імпринтингу. Відповідно з результатами молекулярного аналізу у таких випадках діагноз або підтверджується або спростовується, що є приводом для диференційної діагностики. Молекулярне дослідження також рекомендовано при необхідності визначення батьківського походження мікроделеції, виявленої на попередніх етапах обстеження та при плануванні наступної вагітності в родині, яка має дитину з ССА. Отже, розроблений нами алгоритм дозволяє вибрати оптимальні шляхи проведення генетичного обстеження пацієнтів з підозрою на ССА та може бути використаний для визначення подальшої стратегії медико-генетичного консультування родин, що мають дитину з ССА. На основі одержаних результатів та враховуючи рекомендований нами алгоритм, було розроблено диференційно-діагностичний підхід до формування груп пацієнтів з підозрою на ССА з метою індивідуалізованого та цілеспрямованого використання цитогенетичних, молекулярно-цитогенетичного та молекулярно-генетичного методів. З 78 обстежених пацієнтів ми сформували три групи. До першої групи увійшли пацієнти з наступними синдромами: Вольфа-Хіршхорна, „котячого крику”, Сміта-Мадженіса та „котячого ока”. При діагностиці цих нозологічних форм ХП економічно та стратегічно виправданим є застосовування стандартного цитогенетичного методу. У разі, якщо делецію/дуплікацію не було виявлено, рекомендуємо одразу застосовувати молекулярно-цитогенетичний метод з використанням локус-специфічних зондів на відповідні критичні ділянки. Високочутливий цитогенетичний метод у даної групи пацієнтів може бути застосований при необхідності уточнення точок розривів-з’єднання хромосомного матеріалу, визначеного при стандартному методі. До другої групи увійшли пацієнти з підозрою на синдром Вільямса-Бойрена та синдром мікроделеції 22q11.2. Для верифікації діагнозу у пацієнтів з цієї групи ми рекомендуємо одночасне застосування стандартного цитогенетичного та молекулярно-цитогенетичного методів діагностики ХП. Такий підхід дає можливість прискорити верифікацію діагнозу, що найбільш важливо при необхідності термінового кардіо-хірургічного втручання. До окремої третьої групи увійшли пацієнти з підозрою на синдром Прадера-Віллі, для яких верифікація діагнозу є складною, багатоступеневою і потребує поступового залучення використаних нами методів діагностики. Для встановлення остаточного діагнозу ми рекомендуємо одночасно застосувати стандартний цитогенетичний та молекулярно-цитогенетичний методи діагностики на першому етапі верифікації діагнозу СПВ, та, якщо делецію/мікроделецію критичної ділянки хромосоми 15 не виявлено, продовжити дослідження із використанням методів молекулярного аналізу. ВИСНОВКИ
В дисертаційній роботі вперше запропоновано новий підхід щодо оптимізації генетичної діагностики ССА, який базується на визначенні інформативності стандартних та високочутливих цитогенетичних і молекулярно-цитогенетичного методів та створенні алгоритму генетичного обстеження пацієнтів з підозрою на ССА. Показно гетерогенність прояву кожного із досліджених синдромів як за клінічними, так і за цитогенетичними характеристиками, і необхідність застосування комплексу лабораторних генетичних підходів для підтвердження або спростування попереднього клінічного діагнозу. 1. Доведено необхідність застосування стандартного цитогенетичного методу на першому етапі верифікації діагнозу ССА, який дозволив підтвердити попередній клінічний діагноз ССА у 8,97% пацієнтів, виявити інші хромосомні перебудови у 3,8% пацієнтів, встановити додатковий діагноз – синдром трисомії за довгим і, частково, коротким плечем хромосоми 12, який не відноситься до групи ССА. Встановлено батьківське походження дериватних хромосом у 33,3% випадків та обгрунтовано необхідність цитогенетичного аналізу у батьків при виявленні змін каріотипу у їхньої дитини. 2. Показано, що застосування високочутливого цитогенетичного методу дозволяє виявляти мікроделеції (3,8%) та конкретизувати результати стандартного цитогенетичного аналізу і точки розривів при структурних перебудовах хромосом. 3. Визначено високу специфічність FISH-методу для верифікації діагнозу ССА, застосування якого дозволило підтвердити попередній клінічний діагноз у 86% пацієнтів з синдромом Вільямса-Бойрена, у 47% – з синдромом Прадера-Віллі та 42% пацієнтів з синдромом мікроделеції 22q11.2; спростувати діагноз у 14% пацієнтів з синдромом Вільямса-Бойрена та 58% пацієнтів з синдромом мікроделеції 22q11.2, а також провести диференційну діагностику між синдромом Вільямса-Бойрена (МІМ 194050) та спадковим судинним захворюванням – ізольованим надклапанним стенозом аорти (МІМ 185500). 4. Вперше в Україні та вдруге в світі виявлено поєднання у одного пацієнта двох синдромів: синдрому мікроделеції 22q11.2 та синдрому трисомії за довгим і, частково, коротким плечем хромосоми 12. За допомогою поетапного застосування стандартного цитогенетичного методу та FISH-методу обгрунтовано доцільність одночасного використання обох методів при підозрі на ССА та виявленні інших структурних перебудов в каріотипі пробанда. 5. Встановлено високу інформативність FISH-аналізу із застосуванням локус-специфічних ДНК-зондів на інтерфазних ядрах при відсутності мітотичних хромосом на препаратах лімфоцитів периферичної крові (5,1%). 6. Визначено, що рівень інформативності застосування стандартного цитогенетичного та високочутливого цитогенетичного методів для підтвердження попереднього клінічного діагнозу синдромів: Вольфа-Хіршхорна, „котячого крику”, Сміта-Мадженіса та „котячого ока” склав 100%, синдрому мікроделеції 22q11.2 – 3,85%, в той же час як для синдромів Вільямса-Бойрена та Прадера-Віллі – 0%. Загальна інформативність цитогенетичних методів в нашому дослідженні склала 12,8%. 7. Встановлено, що загальна інформативність молекулярно-цитогенетичного методу для підтвердження або спростування попереднього клінічного діагнозу ССА в нашому дослідженні склала 78,2%, що обґрунтовує доцільність використання FISH-аналізу для верифікації діагнозу синдромів з групи ССА. 8. Визначено, що застосування цитогенетичних, молекулярно-цитогенетичних та молекулярних методів дозволило верифікувати діагноз у 93,6% пацієнтів з підозрою на ССА, на підставі чого розроблено комплексний підхід щодо оптимізації індивідуалізованої діагностики кожного синдрому з групи ССА, який включає формування диференційно-діагностичних груп пацієнтів та створення алгоритму генетичного обстеження пробандів та їх родичів.
Практичні рекомендації
Для лікарів-генетиків, неонатологів, педіатрів, кардіологів, кардіохірургів, дитячих неврологів, імунологів: 1. Направляти на цитогенетичний та молекулярно-цитогенетичний аналіз пацієнтів з характерними для синдромів з групи ССА вродженими вадами розвитку та вродженими вадами серця з метою підтвердження/спростування попереднього клінічного діагнозу. 2. Верифікацію діагнозу у дітей із підозрою на ССА доцільно проводити за запропонованим алгоритмом генетичного обстеження пацієнтів з даною патологією. 3. Виявлення у пробанда структурних перебудов хромосомного матеріалу за участю критичних ділянок хромосом 4, 5, 7, 15, 17, 22 або за участю інших хромосом є підставою для цитогенетичного дослідження лімфоцитів периферійної крові його батьків. 4. У всіх пацієнтів з надклапанним стенозом аорти рекомендовано проводити FISH-аналіз з метою підтвердження або спростування синдрому Вільямса-Бойрена. 5. Виявлення у плоду під час пренатального УЗД вад серця, що характерні для ССА обґрунтовує необхідність проведення пренатальної молекулярно-цитогенетичної діагностики плоду. 6. Наявність мікроделеції/мікродуплікації у пробанда є підставою для проведення пренатальної діагностики в родинах, що мають дитину з певним синдромом з групи ССА.
|
||||
|
|
Последнее изменение этой страницы: 2020-03-26; просмотров: 89; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.119.133.206 (0.007 с.) |