
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Поняття динамічної моделі. Мікро і макрокінетикаСодержание книги
Поиск на нашем сайте
У попередніх розділах ми розглядали процеси, що перебігають в умовах рівноваги, без врахування часу (за деякими виключеннями). В цьому розділі розглянемо процеси, що перебігають у часі – динамічні процеси. До їх числа відноситься більшість хіміко-технологічних процесах. Розділ молекулярної фізики і фізичної хімії, що вивчає перебіг фізичних і хімічних процесів в часі називається кінетикою. Прийнято розрізняти: - фізичну кінетику, що вивчає перебіг процесів, не пов’язаних з хімічними перетвореннями, наприклад перебіг процесів масопереносу, зокрема процеси встановлення рівноваги при фазових переходах; - хімічну кінетику, що вивчає перебіг в часі хімічних реакцій. Наведені вище термодинамічні методи дозволяють розрахувати вихід цільових продуктів хімічних реакцій в умовах рівноваги, але ті термодинамічні результати, що приводять до збільшення виходу продуктів реакції, часто суперечать кінетики процесу. Наприклад, з погляду термодинаміки екзотермічний процес синтезу аміаку бажано вести при низькій температурі, тому що рівноважна ступінь перетворення збільшується при зниженні температури. Однак швидкість реакції, навпаки, при зменшенні температури знижується, тому на практиці треба обирати таку оптимальну температуру Топт., при якій забезпечується максимальний вихід продукту в одиницю часу, тобто максимальна інтенсивність. Тому для встановлення оптимальних технологічних умов необхідно одночасно враховувати як термодинамічні, так і кінетичні фактори. Процеси, що протікають у рідинах і в газах, розглядають з двох точок зору: а) процес взаємодії окремих молекул - на мікрорівні. У системі на мікрорівні рідина являє собою вільні індивідуальні молекули, що рухаються в різних напрямках, що зіштовхуються й змішуються з усіма іншими молекулами даної рідини; б) процес взаємодії агрегатів молекул - на макрорівні. У системі на макрорівні рідина є сукупністю великої кількості маленьких глобул, тобто груп молекул, як би ув'язнених в оболонку. Ця зовнішня оболонка є хімічно інертною. Її призначення полягає в тому, щоб зберегти умовно прийняту індивідуальність кожної глобули. Особливості мікро- і макростану можна простежити при змішанні рідин: чим вище в'язкість рідин, що змішуються, тим яскравіше проявляються в ній ознаки макростану. Як приклад уявимо собі посудину у якиї досить інтенсивно перемішується рідина з малою в'язкістю (вода).
а) у цю рідину вливають іншу рідину теж малої в'язкості (етиловий спирт). Ці рідини добре перемішуються й суміш швидко стає однорідною; умовно можна прийняти, що змішання відбувається на мікрорівні. б) якщо в ту ж посудину замість етилового спирту вливати дуже в’язку рідину (наприклад, гліцерин), а інтенсивність перемішування знизити (зменшити частоту обертань мішалки), гліцерин розіб'ється на окремі краплі.(глобули) і рідина стане однорідної лише після значного проміжку часу. Умовно можна прийняти, що змішання у цьому випадку відбувається на макрорівні. У мікростані рідина не має будь-які властивості, що обумовлені присутністю глобул молекул і, навпаки, властивості рідини, що перебуває в макростані, у значній мірі визначаються наявністю глобул. Реальна суміш у тім або іншому ступені проявляє проміжні властивості, які залежать від властивостей рідини й від системи, у якій відбувається змішання. Всі питання, що стосуються фізичної хімії, - хімічна рівновага, кінетика хімічних реакцій та ін. - розглядаються на мікрорівні, тобто на рівні окремих молекул. Такий підхід дозволяє проаналізувати вплив різних факторів в ідеалізованих умовах. У промислових умовах при оформленні переважної більшості хімічних процесів необхідно враховувати багато супутніх фізичних процесів, що пов'язані з макростаном системи й накладаються на основний хімічний процес. Найважливішими з них є: а) дифузія вихідних реагентів у зону реакції й продуктів реакції із зони реакції, б) виділення й розподіл тепла. На ці процеси сильно впливають аерогідродинамічні умови (тобто характер руху газу або рідини), оскільки від них залежать перенос тепла за рахунок конвекції т й дифузія речовини. Таким чином, при вивченні реального хіміко-технологічного процесу необхідно враховувати вплив дифузії, теплопередачі й конвекції, тобто процес варто розглядати на макрорівні. Швидкість хімічної реакції Хімічні реакції вивчають на основі залежності концентрацій реагентів (у кмоль/м3) і продуктів від часу. Такі залежності носять назви «кінетичні криві». Вони можуть мати різні форму (рис. 14.1):
а) для вхідних продуктів спостерігається тільки зменшення концентрації з часом (рис. 14.1, крива 1);
Рисунок 14.1 – Деякі типові кінетичні криві: 1 – для початкових продуктів, 2 – для кінцевих продуктів, 3 – для проміжних продуктів.
б) для кінцевих продуктів спостерігається тільки монотонне зростання концентрації (рис. 14.1, крива 2); в) для деяких речовин спостерігається спочатку накопичення, але потім – витрата (рис. 14.1, крива 3). Це характерно для проміжних продуктів, які у складних реакцій спочатку утворюються, а потім витрачаються на створення кінцевих продуктів. Швидкість хімічної реакції, що протікає при постійному об’ємі, визначається, як похідна до кінетичної кривої у заданої точці часу:
Швидкість реакції залежить від багатьох причин. На неї впливають: - природа й концентрація реагентів, - тиск (для реакцій за участю газів), - температура, каталізатор, домішки і їхні концентрації, - ступінь здрібнювання (у реакціях за участю твердих речовин), - середовище (для реакцій у розчинах), - форма посудини (у ланцюгових реакціях), - інтенсивність світла (у фотохімічних реакціях), - потенціал електродів (в електрохімічних реакціях), - потужність дози випромінювання (у радіаційно-хімічних процесах). Лише деякі з факторів, що діють на швидкість реакції, одночасно впливають на хімічну рівновагу. У зв'язку із цим треба відзначити величезні труднощі обліку дії різних факторів на швидкість реакції й тим більше кількісної їхньої оцінки. Швидкість процесів контролюється по зміні концентрації реагентів. За їхнім темпом можна стежити й по зміні якої-небудь властивості (показники переломлення, електропровідності, спектра й т.д.), не порушуючи протікання даної реакції. Якщо процес протікає повільно, то через певні проміжки часу відбирають проби й визначають у них зміст даної речовини (наприклад, титруванням). Для. реакцій, що протікають із великою швидкістю, користуються різким охолодженням, зупиняючи їх. Оскільки швидкість реакції дуже чутлива до зміни температури (див. 14.4), суміш що реагує поміщають у термостат, де точно контролюють температуру. Основними параметрами, які доводиться враховувати майже у всіх процесах, є: - концентрації (тиск) реагентів, - температура, - наявність і кількість каталізатору. Вплив концентрації в гомогенних і гетерогенних реакціях. На відмінність від гомогенних процесів, що характеризуються взаємодією речовини в одній фазі, в гетерогенних реакціях поряд з хімічними перетвореннями наявні стадії переносу речовин. Їхній вплив на процес у цілому залежить від умов його протікання: а) якщо найбільш повільною стадією є хімічна реакція, то говорять, що процес протікає в кінетичній області, б) якщо ж, навпаки, ланкою, що гальмує процес у цілому, служить перенос речовин, то говорять що процес протікає в дифузійній області. Яка стадія є такою, що лімітує: взаємодію речовин - можна встановити по температурній залежності швидкості реакції: у першому випадку швидкість набагато дошкульніше до температури, чим у другому. Прості й складні реакції. Якщо процес протікає в одну стадію у відповідності зі стехіометричним рівнянням, тобто відповідає одному етапу, його називають простим.
Складні реакції - це сукупність простих реакцій. До складних реакцій ставляться: - оборотні реакції, - паралельні реакції, - послідовні реакції. Вони розрізняються по характері зв'язку між окремими етапами взаємодії. Для кожної стадії застосовні рівняння простих реакцій. Якщо темп протікання окремих стадій сильно розрізняється, то сумарна швидкість процесу визначається (лімітується) темпом самої повільної стадії й може бути описана кінетичним рівнянням цієї простої реакції. Оборотні реакції характеризуються тим, що протікають в прямому і зворотному напрямках, що забезпечує динамічну рівновагу хімічних реакцій (). Критерієм того, що реакція оборотна, є наявність граничної концентрації продуктів і вхідних речовин від часу, тобто створення рівноважної суміші, склад якої не змінюється з часом. Послідовні реакції характеризуються послідовним створенням і витратою продуктів, аж до створення кінцевого продукту. Приклад:
А + В ® С®D (14.2)
Послідовні етапи реакції, що пов’язані зі створенням речовин С і D називаються стадіями реакції. Критерієм того, що речовина С є проміжним продуктом є наявність максимуму її концентрації на кінетичної кривої (рис. 14.1). Паралельні реакції характеризуються одночасним створенням з одного продукту двох або більше кінцевих продуктів однакової або близької природи. Приклад: при взаємодії толуолу з азотною кислотою супроводжується створенням трьох органічних продуктів – 2-, 3- і 4- нітротолуолів:
Відмітимо, що воду, яка також створюється в цій реакції, не можна вважати за продукт паралельної реакції, це – другій продукт, який необхідно створюється при створені будь якого з ізомерних нітротолуолів. Один з можливих критеріїв паралельної реакції – однакове співвідношення усіх продуктів протягом усій реакції. Окремі прості реакції, сукупність яких створює складну реакцію, називаються елементарними стадіями. Сукупність усіх елементарних стадій та їх швидкостей називається механізм реакції. В лекції вказувалось, що у відношення до швидкостей реакції виконується закон діючих мас: швидкість реакцій є пропорційною добутку концентрацій вхідних реагентів у деяких ступенях, тобто, виконується мультиплікативний закон:
де n1,n2,… - часткові порядки реакції по реагентам 1,2,… Їх сума дає загальний порядок реакції, k – коефіцієнт пропорційності, який має назву «константа швидкості» Знак швидкості: «плюс» - для продуктів реакції, «мінус» - для вхідних речовин.
Швидкість реакції і константа швидкості є величинами розмірними. З рівняння (14.4), оскільки концентрація вимірюється в кмоль/м3, то швидкість в системі СІ вимірюється в Рівняння (14.4) виконується не завжди. Але для елементарних реакцій воно є справедливим завжди, причому у рівняння швидкості реагенти входять у ступенях, які дорівнюють її стехіометричним коефіцієнтам. Це можна використовувати, як правило для укладання рівнянь швидкості окремих реакцій, виходячи з елементарних, що використовується при моделюванні кінетики (див. 14.3) На практиці рівняння (14.4) зручно використовувати, як емпіричну залежність для прикладних розрахунків швидкості. Якщо концентрації усіх реагентів однакові, то швидкість витрати можна розраховувати за спрощеним рівнянням:
де емпіричний порядок n не обов’язково є ціле число. Цей порядок може бути дробовим і навіть негативним. При цьому величина константи швидкості, в загалі кажучи, становиться функцією початкової концентрації реагенту і каталізаторів. Подібні величини в кінетиці називають ефективними константами швидкості. Приклад 14.1 Для деякої реакції A ® B+C були експериментально одержані кінетичні дані, що наведені у табл. 14.1.
Таблиця 14.1 – Залежність концентрацій речовини А в реакції A ® B+C від часу при різних початкових концентраціях
Користуючись цими даними, розрахувати початкову швидкість і визначити її залежність від концентрації реагенту А (винайти істинний порядок реакції) Розв’язання Початкова швидкість – це швидкість реакції у момент часу t = 0:
Для розрахунків початкової швидкості необхідно: ü за експериментальними даними одержати емпіричне рівняння залежності концентрації вхідної речовини або продукту від часу, ü продиференціювати цю залежність, ü розрахувати значення похідної у точці часу t=0. Практика показує, що на початкових ділянках «простих» кінетичних кривих (тобти, таких, що не мають точок перегину) залежність концентрації від часу вдається задовільно описати поліномом 2-го порядку:
C(t) = a0 + a1∙t + a2∙t2. (14.7)
Звідси, після диференціювання:
Для знаходження коефіцієнтів використаємо метод найменших квадратів (функцію LINEST()). Проект з розрахунків наведено на рис. 14.2. За результатами розрахунків будуємо залежність логарифма модуля швидкості від логарифма початкової концентрації. Як випливає з графіка на рис. 14.2, ця залежність є лінійною. Тангенс куту нахилу n = 1,05» 1. Це свідчить, що концентраційний (істинний) порядок реакції дорівнює 1.
Відмітимо, що порядок реакції, що розрахований з часової залежності концентрації (він називається часовим порядком) не обов’язково збігається з концентраційним порядком. Наприклад, спостерігаються випадки, коли у реакції, разом з основним процесов, перебігають побічні, продукти яких гальмують (інгібірують) основний процес. У цьому випадку концентраційний порядок може суттєво відрізнятися від часового, причому, концентраційний порядок характеризує основну реакцію, що не ускладена побічними процесами.
Рисунок 14.2 – Проект «Розрахунок початкових швидкостей»
Константа швидкості (як такі або ефективні) сильно залежать від температури. У 19 в. Вант-Гоф встановити, що при підвищенні температури на 10 градусів константи швидкості збільшуються у 2-4 рази (правило Вант-Гофу). Кількісно для описання залежності констант швидкості від температури частіше за все використовують рівняння Ареніуса:
де постійна А називається передекспоненнціальним множником, постійна Е називається енергією активації. Зміст поняття «енергія активації». Це – додаткова енергія, яку необхідно додати до молекул, що реагують, щоб реакція між ними стала можливою. Для того, щоб молекули прореагували, необхідно, щоб вони достатньо зблизилися. Але при цьому виникають електростатичні сили відштовхування, на подолання яких треба витратити енергію, яку молекули отримують із зовні шляхом нагрівання. Схематично можна зобразити зміну внутрішньої енергії системи у реакції А + В = С на енергетичної діаграмі (рис. 14.3) – залежності внутрішньої енергії системи від координати реакції r (іі в грубому наближенні можна вважати за відстань між реагуючими молекулами). По мірі зближення відбувається зростання енергії за рахунок сил відштовхування між молекулами. Але, водночас, відбувається поступове зав’язування нових зв’язків, яке компенсує це відштовхування. Внаслідок цього у найвищої точці (вона називається перехідним станом або активованим комплексом) встановлюється рівновага сил відштовхування і тяжіння, і потім – створення кінцевих продуктів, що супроводжується зменшенням енергії. При цьому: Е¹ - ЕА+В – це енергія, яку не обходимо додати системі, щоб піднятися до активованого комплексу (енергія активації, тобто енергія, яку необхідно додати, щоб подолати активаційний бар’єр), ЕА+В - ЕС – це енергія, що виділяється під час протікання реакції. Відмітимо, що рис. 14.3 виконано для зручності з порушенням масштабу: як правило енергія активації за величиною менше енергії, що виділяється в реальних хімічних реакціях. Непоганою аналогією до енергії активації може служити процес злипання двох пластилінових кульок: для створення єдиної великої кульки слід спочатку прикласти зусилля для їх здавлювання. Хімічні реакції часто проводіть у присутності каталізаторів, речовин, що прискорюють хімічні реакції, але самі при цьому не витрачаються. Загальна спрощена схема дії каталізаторів така: вони створюють з одним з реагентів (А) нестійку сполуку АС (комплекс реагент-каталізатор), що здатна швидко реагувати з іншим реагентом. При цьому створюється продукт і регенерується каталізатор:
Рисунок 14.3 – Енергетична діаграма хімічної реакції А+В = С
Внаслідок цього каталізатори не впливають на термодинаміку хімічних реакцій. Але їх вплив на кінетику є вирішальним, бо за рахунок створення нестійких реакційно здатних проміжних сполук суттєво знижується енергія активації і збільшується швидкість. Тому концентрація каталізатору завжди входить до складу рівнянь швидкості. Але, оскільки концентрація каталізатору по ходу процесу не змінюється, її можна вносити до складу ефективної константи швидкості реакції. Приклад 14.2 Для реакції 2-го порядку А + В = C + D знайдені такі величини констант швидкості в залежності від температури:
Розрахувати енергію активації реакції. Оцінити погрішність величини енергії активації, вважая розподіл погрішностей нормальним (гауссовим). Оцінити, яким буде час напівперетворення реакціх при температурі 125оС при початкової концентрації реагентів А і В 1,1 кмоль∙м-3. Розв’язання Для розрахунів енергії активації прологарифмуємо формулу (14.9):
З рівняння (14.11) випливає лінійна залежність між логарифмом константи швидкості і зворотною абсолютною температурою 1/Т. При цьому кутовий коефіцієнт дорівнює (-E/R). Для знаходження енергії активації використаємо метод найменших квадратів. Розрахунки виконуємо у середовищі Ооо Calc (рис. 14.4). Для обчислення енергії активації будуємо таблицю вхідних даних, у якої розраховуємо величини 1/Т і ln k. Формули перших комірок таблиці:
D4: =1/(B5+273); E4: =LN(C5)
За даними таблиці будуємо графік. Зі сріншоту випливає, що, дійсно, залежність між ln k та 1/Т є лінійною у нашому випадку. Оскільки необхідно розрахувати не тільки енергію активації але також погрішність цей величини, для розрахунків використовуємо функцію LINEST(), що дає, як вихідні змінні, середні квадратичні (стандартні) відхилення коефіцієнтів лінійної залежності. Формула масиву Н4:І9 буде:
=LINEST(E5:E9;D5:D9;1;1)
У другому рядку масиву містяться шукані величини стандартних відхилень коефіцієнтів. Для розрахунків погрішності у визначення енергії активації використовуємо відому формулу математичної статистики:
де dх – погрішність у визначенні величини х, sх – стандартне відхилення величини х, n – кількість точок,
Рисунок 14.4 – Проект «Розрахунок енергії активації»
Т(nв,a) – критичне значення зворотної нормованої функціі розподілу похибок в залежності від кількості ступенів волі nв і рівня значущості a. Із статистики відомо: в разі того, що похибка у генеральній сукупності розподілена за нормальним законом, те у невеликих вибірках буде спостерігатися розподіл Стьютента. Кількість ступенів волі на 1 менше за кількість точок. У комірці Н15 розрахувуємо критичне значення розподілу Стьюденту. Формула комірки: =TINV(0,05;H14). Далі, шляхом множення кутового коефіцієнту та його похибки на величину газової постійної R, знаходимо енергію активації та її погрішність. Коректне представлення величини енергії активації з врахуванням абсолютної погрішності: Е = (80,0±1,0)∙103 Дж∙моль-1. Виходячі з величини енергії активації та логарифма перед експоненти, розраховуємо величину константи швидкості прі температурі 125оС за рівняннями (14.11) та (14.9). Формули комірок: Н22: =I4+H4/(125+273); Н23: =EXP(H22
Періодом напівперетворення називається проміжок часу від початку реакції до моменту, коли буде вичерпано половину від кількості реагенту. Запишемо рівняння швидкості для реакції 2-го порядку:
При інтегруємо це рівняння аналітично метолдом розділення змінних:
За визначенням:
Звідси знаходимо шуканий період напівперетворення:
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2017-02-10; просмотров: 145; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.142.212.193 (0.011 с.) |


 ; (14.1)
; (14.1)
 , (14.4)
, (14.4)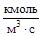 , або, в лінійному запису: кмоль×м-3×с-1. Для зручності прийнято при виконанні розрахунків переходити від секунд до більш крупних одиниць часу: хвилин або годин. Для реакції n-го порядку у правої частині буде стояти вираз Сn, тобто кмольn×м-3n. Звідси розмірність константи швидкості складає кмоль(1-n)×м-3(1-n)×c-1
, або, в лінійному запису: кмоль×м-3×с-1. Для зручності прийнято при виконанні розрахунків переходити від секунд до більш крупних одиниць часу: хвилин або годин. Для реакції n-го порядку у правої частині буде стояти вираз Сn, тобто кмольn×м-3n. Звідси розмірність константи швидкості складає кмоль(1-n)×м-3(1-n)×c-1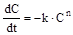 . (14.5)
. (14.5) (14.6)
(14.6) (14.8)
(14.8)
 , (14.9)
, (14.9)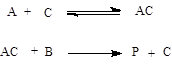

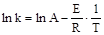 . (14.11)
. (14.11) , (14.12)
, (14.12)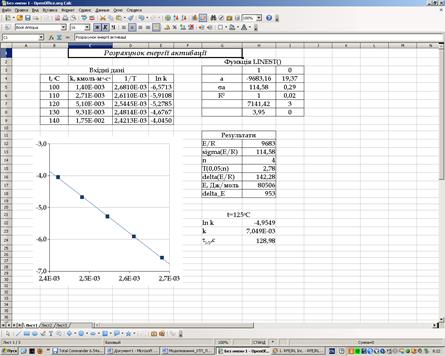
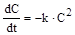 , С(t=0)=C0 (14.13)
, С(t=0)=C0 (14.13) ;
;  . (14.14)
. (14.14)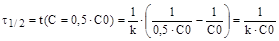 . (14.15)
. (14.15) с
с


