
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Кондуктометричні методи аналізу
4.3.1. Електропровідність розчинів електролітів. Кондуктометричні методи аналізу грунтуються на залежності електричної провідності розчинів електролітів від їх складу. Електричною провідністю називають здатність речовин проводити електричний струм під дією зовнішнього джерела електричного поля. Носіями зарядів в розчинах електролітів є іони різного заряду: катіони і аніони, які рухаються відповідно до електродів з протилежним зарядом. Для вимірювання електричної провідності в розчин занурюють два індиферентних електрода, до яких прикладають напругу від джерела електричного струму. Для запобігання зміни складу розчину завдяки електролізу і зменшенню поляризації електродів при вимірюванні електропровідності розчинів електролітів використовують змінний струм. Електрична провідність вимірюється як сила струму, яка проходить через розчин в розрахунку на одиницю прикладеної до електродів напруги, і розраховується за формулою:
де W – електрична провідність, См; І - сила струму, А; U - напруга, В. Одиницею електричної провідності є Сіменс (См). Електрична провідність обернено пропорційна опору:
розмірність електричної провідності [См] = [Ом-1]. Електрична провідність розчинів електролітів залежить від розмірів і розташування електродів, температури, природи розчинника, властивостей іонів і їх концентрації. Електрична провідність прямопропорційна площі електродів (S) і обернено пропорційна відстані між електродами (l):
Коефіцієнт пропорційності æ називається питомою електричною провідністю і дорівнює електричній провідності розчину при вимірюванні з допомогою електродів площею 1 см2, які розташовані на відстані 1 см один від одного:
Відношення l/S називають константою електролітичної комірки. Питома електрична провідність залежить від температури, розчинника, властивості йонів і їх концентрації. Для дисоціації AB ↔ Az+ + Bz-:
де a - ступінь дисоцації електроліта. Для сильних електролітів a ~ 1, С - молярна концентрація електроліта, F - число Фарадея, z +, z - - заряд катіона та аніона, u +, u - - швидкість руху катіона та аніона при напруженості електричного поля 1 В/см. Питома електрична провідність, віднесена до числа моль-еквівалентів іонів в 1 см3 розчину, називається еквівалентною електричною провідністю або рухливістю (l):
де С - концентрація йонів, моль-еквівалентів/літр, V – розбавлення – це об’єм розчину в см3, який при відстані між електродами 1 см містить 1 моль еквівалентів іонів. У кондуктометричних методах аналізу вимірюваним параметром, який залежить тільки від складу розчину електроліту, є питома електрична провідність (æ) або електрична провідність (W), виміряна при незмінній константі електрометричної комірки (l/S). Константу електрометричої комірки зазвичай розраховують за формулою 4.31, вимірявши електричну провідність стандартного розчину електроліту з відомою питомою електричною провідністю. Електричну провідність вимірюють кондуктометрами або розраховують за формулою 4.29, вимірявши опір між електродами (R) реохордним містком. В обох випадках до електродів прикладається змінна напруга. Під час вимірювань обидва електроди повинні повністю бути занурені в розчин. Питома та еквівалентна електрична провідності залежать від властивості іонів та їх концентрації, температури і розчинника. При низьких концентраціях електролітів зростання концентрації призводить до збільшення кількості носіїв заряду, що пропорційно збільшує електропровідність (рис. 4.10,а).
Рис. 4.10. Залежність питомої електричної провідності (а) і еквівалентної електричної провідності (б) від концентрації електролітів (1 – HCl, 2 – KOH, 3 – CH3COOH).
При збільшенні концентрації зростання електропровідності відстає від лінійної залежності, що для слабких електролітів пов'язане зі зменшенням ступеню дисоціації (a), а для сильних електролітів - із збільшенням іонної сили розчину. При великих концентраціях посилюється міжіонна взаємодія і швидкість руху іонів продовжує зменшуватися внаслідок катафоретичного і релаксаційного ефектів. Катафоретичний ефект полягає в тому, що атмосфера протийонів, яка оточує кожний іон, рухається в протилежному напрямку і гальмує рух іону. Релаксаційний ефект пояснюється тим, що при русі іону його іонна атмосфера руйнується і створюється на новому місці. Ці ефекти призводять до того, що не тільки уповільнюється зростання електричної провідності, але інколи електрична провідність зменшується зі зростанням концентрації.
Збільшення концентрації електролітів призводить до зменшення еквівалентної електроровідності розчину (рис. 4.10,б). Для сильного 1-1 валентного електроліту її можна розрахувати за рівнянням Онзагера:
де A - константа, яка залежить від температури, в’язкості розчину і діелектричної проникності розчинника, m - йонна сила розчину. l 0 - гранична еквівалентна електрична провідність (гранична рухливість) - еквівалентна електрична провідність при безмежному розведенні. Гранична рухливість залежить від температури і природи розчинника. Вона визначається екстраполяцією залежності еквівалентної електропровідності від концентрації електроліта до перетину з віссю ординат. Кольрауш встановив, що гранична еквівалентна електропровідність розчину електроліту є адитивною величиною і складається з граничних рухливостей катіонів і аніонів: l 0 = l + + l – (4.35) Гранична еквівалентна електрична провідність є фізико-хімічною константою іонів у певному розчиннику при певній температурі і наводиться у довідниках. Граничні рухливості різних іонів у водних розчинах для більшості іонів знаходяться в межах 40 – 70 См·см2. Граничні рухливості H+ та OH– дорівнюють 362 і 205 См·см2 відповідно, що сильно відрізняється від рухливості інших іонів. Ця аномалія пояснюється естафетним механізмом передавання заряду цими іонами з утворенням водневих зв’язків:
або H – O– + H – O – H = │H – O .. H .. O – H│– = H – O – H + –O – H. З адитивності рухливостей і формули (4.33) можна вивести формулу для питомої електричної провідності розчину, який містить декілька іонів:
Електрична провідність і рухливість збільшуються зі збільшенням температури, бо зменшується в’язкість розчинника і збільшується швидкість теплового руху іонів:
де α - температурний коефіцієрт електричної провідності, який залежить від природи іонів і розчинника, t - температура розчину, оС. Для водних рочинів цей коефіцієнт дорівнює 0,02 – 0,025. Для точних вимірювань, щоби виключити вплив температури, електрометричну комірку термостатують. Електрична провідність і рухливість залежать від природи розчинника. А.М. Шкодіним встановлена залежність граничної рухливості від діелектричної проникності і в’язкості розчинника:
де A i B - константи, h – в’язкість розчинника, D - діелектрична проникність розчинника. В неводних розчинах у більшості іонів рухливість менша ніж у водних. Аналітичний сигнал кондуктометрії (W або æ) є одномірним і неселективним, його величина залежить від природи і концентрації всіх іонів, які знаходяться в розчині (формула 4.36). Тому якісний аналіз цим методом проводити не можна.
4.3.2 Пряма кондуктометрія. Кількісний аналіз методом прямої кондуктометрії проводиться в таких випадках: а). Аналіз бінарних розчинів. Бінарні розчини складаються з двох компонентів - розчинника і одного розчиненого електроліта. Наприклад, вода-H2SO4, вода-NaCl, H2SO4 -SO3 (олеум). б). Аналіз псевдобінарних розчинів. Псевдобінарні розчини містять декілька електролітів, але за умовами їх використання змінюється концентрація одного електроліта, а концентрації інших електролітів постійні. Наприклад, водний розчин NaCl - NaOH.
в). Контроль чистоти розчинника. Якщо чистий розчинник має низьку електричну провідність, наприклад, дистильована вода, а домішки електролітів електричну провідність різко збільшують, значення питомої електричної провідності характеризує чистоту розчинника. У випадках а) і б) визначення концентрації електроліта проводять методом абсолютного калібрування, і тільки у випадку б) стандартні розчини необхідно готувати з врахуванням постійних концентрацій інших електролітів. У випадку в) заздалегідь визначають, яка питома електрична провідність відповідає необхідній чистоті розчинника. Очистку проводять доти, поки питома електрична провідність очищеного зразка стане меншою визначеного значення. Цей варіант можна використати для контролю очистки стічних вод або для приблизного визначення загальної мінералізації технологічних і природних вод. Переваги метода прямої кондуктометрії: простота, надійність, достатня точність результатів (1-2%), доступне обладнання. Недоліки: обмеженість об’єктів аналізу, вплив на результати забруднення сторонніми електролітами; при високих концентраціях результати можуть бути не однозначними.
4.3.3. Кондуктометричне титрування. Ширше застосування дістав метод кондуктометричного титрування, який полягає в побудові залежності електричної провідності розчину від об’єму титранта (кривої титрування) і визначенні точки еквівалентності за зламом на кривій титрування. Розчин, який аналізують, вміщають в електрометричну комірку з перемішуючим пристроєм (магнітною мішалкою). Розчин титранта однаковими порціями додають в комірку і через деякий час записують електричну провідність розчину. Якщо титрування триває декілька хвилин, комірку можна не термостатувати. Для одержання точніших результатів і при тривалому титруванні електрометричну комірку термостатують. Щоб зменшити зміну електричної провідності розчину за рахунок розбавлення його титрантом, бажано використовувати титрант з більшою концентрацією, який подають з мікробюретки. Злам на кривій титрування спотерігається, якщо одним з продуктів реакції титрування є малодисоційована сполука або відбувається перетворення іонів в молекулярну форму і навпаки. Кислотно-основне титрування. При титруванні сильних і слабких кислот та основ малодисоціюючим продуктом є вода. Наприклад, для сильної кислоти:
H+ + An- + Kt+ + OH- = Kt+ + An- +H 2 O До точки еквівалентності в розчині відбувається заміна рухливих іонів водню на малорухливі іони Kt+ і електрична провідність зменшується. Після точки еквівалентності продовжується збільшення концентрації іонів Kt+ і з’являється надлишок іонів гідроксилу, що призводить до збільшення електричної провідності. При титруванні слабких кислот, в залежності від силового показника кислоти, зменшення електричної провідності до точки еквівалентності буде не таким різким, як для сильних кислот, але після точки еквівалентності зростання кривої буде таким самим (рис. 4.11 (а, б, в)).
Рис. 4.11. Криві кондуктометричного титрування сильної кислоти (а), кислоти середньої сили (б), слабкої кислоти (в), суміші сильної і слабкої кислоти (г).
При титруванні суміші слабкої і сильної кислоти можливе фіксування двох точок еквівалентності, якщо різниця силових показників кислот перевищує 4 (рис. 4.11г). Спочатку титрується сильна кислота до першої точки еквівалентності. Витрата титранта між першою і другою точкою еквівалентності відповідає титруванню слабкої кислоти. Метод осадження. При утворенні малорозчинних сполук до точки еквівалентності теж відбувається заміна одних іонів іншими при однаковій їх концентрації. Наприклад: Na+ + Cl- + Ag+ + NO3 - = AgCl ↓ + Na+ + NO 3- До точки еквівалентності іони хлору замінюються на іони нітрату, які мають дещо меншу рухливість, і електропровідність буде дещо зменшуватись. Після точки еквівалентності, завдяки збільшенню концентрації іонів срібла і нітрату, електропровідність буде суттєво збільшуватись. Якщо в розчині присутні 2 іони, які утворюють малорозчинні сполуки з титрантом і відношення добутків розчинності цих іонів більше 104, то при суттєвій відмінності рухливостей протиіонів теж можлива фіксація двох точок еквівалентності. Метод комплексоутворення. Утворення комплексних сполук супроводжується зменшенням кількості іонів в розчині і, відповідно, зменшенням електропровідності, наприклад: 2 Cl- + Hg 2+ = [HgCl 2 ] Al 3+ + 6 F- = [AlF 6 ] 3- Після точки еквівалентності електрична провідність збільшується через збільшення надлишку титранта. При використанні найбільш широковживаного метода комплексонометрії відбуваються такі реакції: до т.е. Men+ + H 2 Y 2- = [MeY]n -4 + 2 H+, після т.е. H+ + H 2 Y 2- = H 3 Y -. До т.е. кількість зарядів не змінюється, але з’являються більш рухливі іони водню і електрична провідність збільшується. Після т.е. іони водню зв’язуються з надлишком комплексону і електрична провідність зменшується. Якщо комплексонометричне титрування проводять у присутності буферних розчинів, точку еквівалентності можна визначати при відмінності рухливостей іонів, які зв’язуються в комплекс і які накопичуються після точки еквівалентності. Якщо іони реагують з комплексоном при різних значеннях pH, вони можуть бути окремо визначені в одному розчині.
Окисно-відновне титрування. Окисно-відновне кондуктометричне титрування практично неможливе, якщо реакція проводиться в сильно кислому або в сильно лужному середовищі (перманганатометрія, біхроматометрія). У цих випадках розчин має велику електричну провідність, яка в процесі титрування мало змінюється. Якщо ж реакція відбувається з достатньою швидкістю в середовищі близькому до нейтрального, зміна електричної провідності в точці еквівалентності стає достатньою для її фіксування. Наприклад, при йодометричному визначенні арсенітів в присутності соди проходять такі реакції: AsO 32- + I 2 + H 2 O = AsO 43- + 2 H + +2 I - 2 H + + 2 HCO 3- = H 2 CO 3 В результаті першої реакції утворюються рухливі іони I- (l 0 = 79), а іони H+ зв’язують мало рухливі іони HCO3- (l 0 = 44) і до точки еквівалентності електропровідність розчину збільшується. Після точки еквівалентності надлишок титранту (спиртовий розчин І2) не змінює електропровідності. Точність кондуктометричного титрування залежить від характеру зламу кривої титрування в точці еквівалентності. При титруванні сильних кислот та основ до і після точки еквівалентності електрична провідність змінюється практично лінійно, і точка еквівалентності точно визначається за перетином цих прямих. В інших випадках поблизу точки еквівалентності спостерігається більш-менш плавний перехід зумовлений недостатньо великим значенням константи рівноваги реакції титрування. В цьому випадку точку еквівалентності визначають за перетином прямих, проведених через точки досить віддалені від точки еквівалентності. Кондуктометричне титрування легко піддається автоматизації. Зазвичай використовується подача титранта з постійною витратою і фіксуванням кривої титрування на діаграмній стрічці реєстратора. Точка еквівалентності титрування фіксується за часом, який пройшов від початку титруванні до зламу на кривій титрування. Такий варіант аналізу називається хронокондуктометричним титруванням. Перевагою кондуктометричного титрування є можливість проведення аналізу каламутних та забарвлених розчинів, використання різних титриметричних методів, визначення органічних і неорганічних сполук, диференційованного титрування сумішей декількох компонентів без розділення. Нижня межа визначуваних концентрацій - 10-4 моль/л при точності не менше 2 %.
|
|||||||||
|
|
Последнее изменение этой страницы: 2017-02-07; просмотров: 358; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.14.83.223 (0.031 с.) |
 , (4.28)
, (4.28)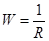 , (4.29)
, (4.29) (4.30)
(4.30) (4.31)
(4.31) (4.30)
(4.30) , (4.33)
, (4.33)
 , (4.34)
, (4.34)
 (4.36)
(4.36) (4.37)
(4.37) , (4.38)
, (4.38)



