Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Нарушения обмена фенилаланина и тирозина
Нарушения обмена этих АК связано с нарушением биосинтеза некоторых ферментов, которые катализируют метаболические превращения этих АК. Результатом нарушения синтеза ферментов является возникновение наследственных генетических заболеваний: 1) фенилкетонурия - нарушен синтез фенилаланин-гидроксилазы, поэтому фенилаланин превращается в фенилпируват, который оказывает токсическое воздействие на развитие некоторых отделов головного мозга. 2) альбинизм - нарушен синтез ферментов, превращающих ДОФА в ДОФА-хром, поэтому нарушается синтез меланинов. 3) алкаптонурия - нарушен синтез диоксигеназы гомогентизиновой кислоты, она выделяется с мочой, моча приобретает черный цвет. 4) кретинизм - нарушен синтез йодиназы, что приводит к нарушению синтеза йодсодержащих гормонов щитовидной железы. 5) может быть нарушен синтез фермента тирозиназы, который катализирует превращение тирозина в ДОФА, следовательно будет нарушаться синтез гормонов мозгового слоя надпочечников и меланина. Из всех этих заболеваний в настоящее время удается лечить фенилкетонурию, для этого из рациона ребенка исключают фенилаланин и увеличивают в пище количество тирозина. Если ребенка держать на этой диете до 6-7 лет, тогда не возникает умственная отсталость, т.к. к 6-7 годам успевают развиться отделы головного мозга, развитие которых задерживается при избытке в ткани мозга фенилпирувата. №24. Метаболические дефекты при классической и атипичной фенилкетонурии. Основные проявления, терапевтическая тактика. Фенилкетонурия (phenyiketonuria; фенилаланин + кетоны + греч. uron моча; синоним: фенилпировиноградная олигофрения, болезнь Феллинга) — наследственная болезнь, обусловленная нарушением обмена фенилаланина; проявляется отставанием в физическом развитии и прогрессирующим слабоумием, расстройствами движений и мышечного тонуса . Средняя частота встречаемости патологии по результатам массовых обследований новорожденных составляет 1:10 000; мальчики и девочки болеют одинаково часто, однако мальчики чаще погибают на 1-м году жизни. Фенилкетонурия Заболевание вызывается отсутствием или дефицитом фенилала-нингидроксилазы и является одним из самых частых нарушений обмена аминокислот.
Фенилаланин в организм в основном поступает с пищей и ката-болизируется в тирозин в печени ферментом фенилаланингидрокси-лазой, содержащей вкачестве кофактора тетрагидробиоптерин. Этот фермент также присутствует в лейкоцитах. Описано много мутаций, вызывающих дефицитфенилаланингидроксилазы. При дефиците фермента возрастает концентрация фенилаланина в крови, активируются альтернативные метаболические пути, в ходекоторых образуются фениллактат, фенилацетат и многие другие метаболиты, в норме определяемые в очень небольших количествах. При фенилкетонурии альтернативные метаболиты экскретируются в мочу, где фениллактат обусловливает характерный запах («запах мышей»), определяемый унелеченых пациентов. Стойкая постнаталь-ная гиперфенилаланинемия вызывает необратимые поражения мозга в результате сложных механизмов, включающихнарушение обмена аминокислот и угнетение синтеза нейромедиаторов. Фенилаланин является конкурентным ингибитором тирозиназы — ключевого ферментасинтеза меланина. Дефицит указанных ферментов вызывает наследственные метаболические заболевания. Клинические проявления Дети с фенилкетонурией нормальны при рождении, так как фенил-аланин быстро переносится через плаценту. При классической фенил-кетонурии накоплениефенилкетонов и нарушение умственного развития проявляются к 6 месяцам после рождения. У некоторых детей появляются судороги, агрессивное поведение,может наблюдаться микроцефалия. Для больных фенилкетонурией характерна тенденция к гипопигментации (вследствие угнетения фенилаланином тирозиназы),что проявляется наличием у детей светлых волос и голубых глаз. Диагностика Диагноз должен быть поставлен, а лечение начато в первый месяц после рождения для предотвращения задержки в умственном развитии. Разработаныскрининговые программы для новорожденных, в частности основанные на применении теста Гатри. Это микробиологический тест, при котором дискфильтровальной бумаги, содержащий кровь из пятки, помещают на чашку с посеянными микроорганизмами, нуждающимися для своего роста в фенилаланине,источником которого является образец крови. Рост микроорганизмов определяется как положительный тест, указывающий на необходимость определенияконцентраций фенилаланина в крови.
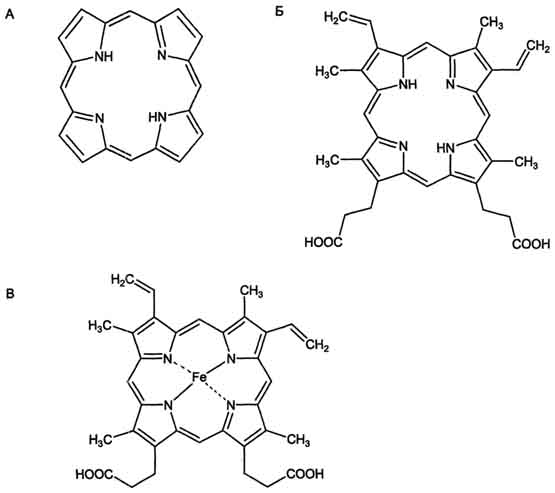
Лечение Лечение состоит в специальной диете, при которой белок замещается смесью аминокислот с низким содержанием фенилаланина. Лечение должнопродолжаться в течение многих лет, так как повышенные уровни фенилаланина между 4 и 8 годами приводят к задержке умственного развития. Ранее полагали,что пищевой контроль (безфенилаланиновая диета) необходим только в течение первых 10 лет жизни, однако современные данные говорят о необходимости пожизненного придерживания этой диеты. В некоторых случаях даже раннее начало лечения не может предотвратить умственные нарушения, но в этих случаях тяжесть их существенно меньшая, чем вотсутствие лечения. Пациенты с фенилкетонурией должны воздерживаться от употребления каких-либо продуктов, содержащих метиловый эфир М-аспар-тилфенилаланина(аспартам) — очень распространенный искусственный заменитель сахара. Эта пищевая добавка расщепляется в ЖКТ до фенилаланина. Особенно важнымявляется то, что все пищевые продукты, в том числе сладкие напитки, должны иметь указание на этикетке о содержании этого искусственного сахара. Другие причины фенилкетонурии Кроме фенилкетонурии и дефицита тетрагидробиоптерина (приводящих к тяжелым клиническим проявлениям), частичный дефицит фенилаланингидроксилазыможет вызывать умеренную или атипичную фенилкетонурию. № 25. Общая схема синтеза гема. Нарушения синтеза гема-порфирии. Интоксикация свинцом. Строение гема Гем состоит из иона двухвалентного железа и порфирина (рис. 13-1). В основе структуры порфиринов находится порфин. Порфин представляет собой четыре пиррольных кольца, связанных между собой метеновыми мостиками (рис. 13-1). В зависимости от структуры заместителей в кольцах пирролов различают несколько типов порфиринов: протопорфирины, этиопорфирины, мезо-порфирины и копропорфирины. Протопорфирины - предшественники всех других типов порфиринов. Гемы разных белков могут содержать разные типы порфиринов (см. раздел 6). В теме гемоглобина находится протопорфирин IX, который имеет 4 метальных, 2 винильных радикала и 2 остатка пропионовой кислоты. Железо в теме находится в восстановленном состоянии (Fe+2) и связано двумя ковалентными и двумя координационными связями с атомами азота пиррольных колец. При окислении железа гем превращается в гематин (Fe3+). Наибольшее количество гема содержат эритроциты, заполненные гемоглобином, мышечные клетки, имеющие миоглобин, и клетки печени из-за высокого содержания в них цитохрома Р450.
Рис. 13-1. Строение порфина (А), протопорфирина IX (Б) и гема гемоглобина (В). Порфин - циклическая структура, состоящая из четырёх пиррольных колец, связанных между собой метановыми мостиками. Протопорфирин IX имеет четыре метильных, два винильных радикала и два остатка пропионовой кислоты. В теме гемоглобина Fe2+ образует две ковалентные и две координационные связи с атомами азота пиррольных колец протопорфирина IX. Гем синтезируется во всех тканях, но с наибольшей скоростью в костном мозге и печени (рис. 13-2). В костном мозге гем необходим для синтеза гемоглобина в ретикулоцитах, в гепатоцитах - для образования цитохрома Р450.
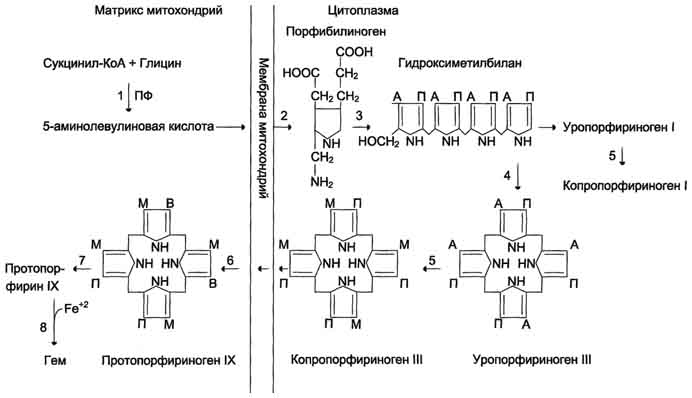
Первая реакция синтеза гема - образование 5-аминолевулиновой кислоты из глицина и сук-цинил-КоА (рис. 13-3) идёт в матриксе митохондрий, где в ЦТК образуется один из субстратов этой реакции - сукцинил-КоА. Эту реакцию катализирует пиридоксальзависимый фермент аминолевулинатсинтаза. Из митохондрий 5-аминолевулиновая кислота поступает в цитоплазму. В цитоплазме проходят промежуточные этапы синтеза гема: соединение 2 молекул 5-аминолевулиновой кислоты молекулу порфобилиногена (рис. 13-4), дезаминирование порфобилиногена с образованием гидроксиметилбилана, ферментативное превращение гидроксиметилбилана в молекулу уропор-фобилиногена III, декарбоксилирование последнего с образованием копропорфириногена III. Гидроксиметилбилан может также нефермента-тивно превращаться в уропорфириноген I, который декарбоксилируется в копропорфирино-ген I. Из цитоплазмы копропорфириноген III опять поступает в митохондрии, где проходят заключительные реакции синтеза гема. В результате двух последовательных окислительных реакций копропорфириноген III превращается в протопорфириноген IX, а протопорфириноген IX - в Протопорфирин IX. Фермент феррохела-таза, присоединяя к протопорфирину IX двухвалентное
|
|||||||
|
|
Последнее изменение этой страницы: 2017-01-24; просмотров: 912; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 13.58.216.18 (0.01 с.) |