
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Обеспечение качества лекарственных средств и медицинских изделий
Госстандарт России в 1997 г. зарегистрировал самостоятельную систему сертификации фармацевтических препаратов и утвердил знак соответствия. Это сделало возможным осуществлять предреализационный контроль всех серий препаратов по производственным протоколам, в которых указываются все стадии производственного процесса, серии, изъятые для контроля, результаты анализа и т.д. Данный контроль позволяет осуществлять профилактику возможных нарушений и стимулировать производство продукции высокого качества. Другой формой контроля качества фармацевтической продукции является сертификация самого производства. Сертификат выдается на выпуск конкретного препарата; проверяется уровень его соответствия требованиям международного стандарта GMP (надлежащая производственная практика) и только после этого выдается сертификат производства. Без этого сертификата фармацевтическая фирма не может получить лицензию на производство и реализацию своей продукции. Эти меры дают определенную государственную гарантию качества производимых препаратов. На рынке фармацевтической продукции сертификат Госстандарта появился в апреле 2000 г., после выхода постановления Правительства РФ о включении ЛС в перечень товаров, подлежащих обязательной сертификации. В соответствии с этим постановлением в Минздраве РФ создана единая информационно-учетная система, в которую заносятся все сертификаты. Сертификат позволяет определить не только маршрут движения ЛС, но и количество ввезенной или произведенной в стране продукции. Органы по сертификации в любом регионе России имеют возможность через компьютерную базу выяснить все интересующие подробности о предъявителе сертификата, а также вести учет объема и номенклатуры лекарственной продукции, находящейся в обороте на фармацевтическом рынке. Работой по недопущению фальсифицированных препаратов на фармацевтический рынок (совместно с правоохранительными органами) и выявлению такой продукции в фармацевтических организациях оптовой и розничной сети и на производственных предприятиях должна заниматься государственная система Службы медицинского и фармацевтического надзора в области обращения ЛС. Для сравнения можно сказать, что персонал в аналогичной Фарминспекции структуры США составляет более 10 тыс. человек. Решение проблемы фальсификации ЛС возможно в рамках формирования национальной лекарственной политики.
В настоящее время надзорная деятельность в области обеспечения контроля качества и безопасности ЛС распространяется пока еще не на все организации и предприятия, производящие и реализующие медицинские препараты. Качество данной продукции не всегда соответствует международным требованиям и стандартам, а также рекомендациям Комитета ВОЗ по биологической стандартизации. На фармацевтических рынках большинства стран мира в сфере обращения находится большое количество малоэффективных, недоброкачественных, небезопасных и фальсифицированных биологически активных пищевых добавок (БАД) и ЛС. Мировой оборот фальсифицированных ЛС и БАД предположительно оценивается в 2,5 млрд долларов в год. Масштабы распространения фальсифицированных ЛС в России разными участниками фармацевтического рынка оцениваются неоднозначно. По данным опроса 2002 г., проведенного среди крупнейших российских и зарубежных фармацевтических компаний, контролирующих свыше 55% объема фармацевтического рынка, каждый 10-й препарат, реализованный на фармацевтическом рынке страны, является подделкой. Ущерб, приносимый этими средствами, по их мнению, составляет более 250 млн долларов в год. Происходящий в последние годы интенсивный рост количества фальсифицированных ЛС создает реальную угрозу здоровью населения. Несовершенство действующего законодательства, регламентирующего фармацевтический рынок, является важным фактором, способствующим массовому распространению фармацевтических фальсификатов. Своего решения требует задача совершенствования нормативно-правовой базы не только по вопросам регистрации ЛС, но и по ее гармонизации с международными требованиями. Необходимо осуществить ряд комплексных мер, связанных с созданием эффективной системы контроля процесса доклинических и клинических исследований, нормативноправовых основ деятельности контрольно-исследовательских организаций. Особое внимание должно быть направлено на обучение персонала через региональные обучающие семинары и широкое использование на практике «Правил организации клинических исследований лекарственных средств (GCP)».
Развитию отечественной фармацевтической отрасли и повышению качества выпускаемой ею продукции будут способствовать следующие мероприятия: • внедрение правил GMP в производство и GLP - в научную практику; • гармонизация требований к проведению клинических исследований в рамках ICH, GCP; • организация и управление системой контроля качества; • восстановление производства фармсубстанций по правилам GMP; • интеграция научного и производственного потенциала фарминдустрии в единую инновационную систему; • создание условий для продвижения ЛС на рынок и функционирования инновационной системы в этом секторе здравоохранения; • организация единой государственной инновационной фармацевтической структуры, объединяющей все стадии разработки и внедрения новых ЛС, а также технологии их производства; • создание наднациональных систем фармаконадзора; • эффективное функционирование экспертного органа по разработке и имплементации (от лат. implere - выполнять) стандартов развития и совершенствования отрасли. Новые социально-экономические и организационно-правовые условия, возникшие благодаря рыночным процессам в стране, оказали влияние на контрольно-разрешительную систему качества продукции в фармацевтической отрасли. К особенностям такого влияния можно отнести: • предоставление аптекам прав на юридическую и экономическую самостоятельность; • активизацию процесса приватизации и развитие частного предпринимательства; • бесконтрольное увеличение числа оптовых и розничных фармацевтических организаций; • присутствие на рынке ЛС большого числа иностранных фирм, имеющих преимущества перед российскими. На российском рынке отечественная фарминдустрия представлена в основном производством малоэффективных ЛС, создававшихся в 50-60-х годах прошлого века. Большая часть наиболее «ходовых» медикаментов госпитальной группы закупается по импорту, причем со сроком годности нередко 3-4 мес за 30-40% от номинала. Из-за сильной конкуренции и перепроизводства только в Европе ежегодно не находит сбыта и утилизируется до 15% лекарственных препаратов. Ежегодно в России регистрируется около 1200 препаратов, в основном генериков. Возникла «фармацевтическая зависимость» России от западных стран: более 60% российского рынка ЛС представлены импортными препаратами. До 70% этого рынка в России приходится на препараты, которые давно не используются в мировой практике из-за малой эффективности и морального старения, недостаточного качества, однако они относительно дешевы, что привычно для стереотипа отечественного потребителя. Обеспечивать выпуск высокоэффективных ЛС позволяет внедрение в производство национальных правил GMP, при этом действуют строгий учет и жесткий документальный контроль каждой стадии технологического процесса. В ближайшие годы необходим перевод предприятий на работу в режиме GMP; такой переход связан со значительными инвестициями. Создание системы управления качеством, производством фармсубстанций по правилам GMP входит в стратегические цели и задачи развития отечественной фармацевтической отрасли. Помимо внедрения правил GMP в производство и реализацию фармпрепаратов остро стоит задача внедрения системы GLP в научную практику.
Важнейшим в обеспечении качества ЛС является этап клинических испытаний нового лекарственного препарата, заключающийся в подборе оптимальных доз и схемы назначения для получения терапевтического эффекта (до полного излечения заболевания или для снятия отдельных его симптомов). В ряде случаев фармацевтические компании уже после выхода препарата на международный рынок вынуждены менять рекомендованные дозы. Подобная практика, по результатам исследований, позволяет скрывать недочеты клинических испытаний. По данным американского Центра разработки и исследования ЛС (подразделения FDA), 21% всех лекарственных препаратов, созданных в период с 1980 по 1999 г., требовал изменения рекомендованных производителями доз. В 80% случаев речь шла о снижении максимальных суточных доз. Причиной коррекции рекомендаций служили многочисленные сообщения о регистрации неописанных производителем побочных эффектов препарата. Наиболее часто изменениям подвергались дозировки препаратов, предназначенных для лечения сердечно-сосудистых заболеваний. Характерна тенденция постепенного роста частоты подобных случаев, кроме того, увеличиваются темпы этого роста. Необходимость в коррекции рекомендуемых доз чаще является следствием ошибок доклинических исследований, а также допущенных при планировании клинических испытаний препарата. Как правило, производители стараются указывать в рекомендациях завышенные дозы, «повышая» таким образом «эффективность» препарата. В результате пациенты получают дозы, граничащие с токсичными, и только когда доходит до опасных побочных эффектов, фармацевты вынуждены признавать свои ошибки. Правда, не установлены побочные эффекты, возникающие из-за конкретной некачественной серии препаратов, поступившей в продажу. Кроме того, побочные эффекты, помимо ошибочного перевода больного с одного препарата на другой, могут быть следствием невыявленных побочных проявлений в период предрегистрационных испытаний. Современные условия позволяют проводить международные мультицентровые испытания не только в клиниках Москвы и Санкт-Петербурга, но и в регионах России, увеличивая таким образом потенциал в проведении будущих клинических испытаний.
Экспертизу эффективности и безопасности ЛС осуществляют экспертные советы. Порядок регистрации фармакологических средств определен соответствующими статьями Закона «О лекарственных средствах» и некоторыми нормативными документами, в частности положениями о фармакологическом и фармакопейном комитетах. Если в советские времена действовала централизованная система производства и реализации ЛС, то в условиях хозяйственной свободы предприятия пошли «своим путем» и стали вносить в фармакопейные статьи сведения об одном и том же препарате, часто исключающие друг друга и вызывающие претензии у потребителей. Теперь в соответствии с мировой практикой и приказом Минздрава РФ «О введении в действие отраслевого стандарта» (2001) каждый производитель отвечает за свою продукцию. При этом сроки проведения экспертизы ЛС (большую часть времени занимают клинические исследования) составляют несколько лет, что тоже соответствует мировой практике. Процедура проведения клинических испытаний требует соблюдения прав пациента, который должен быть застрахован в установленном законом порядке, а также иметь возможность выйти на любом этапе из процесса исследования. Срок годности нормативной документации на все медицинские препараты (отечественные и зарубежные) составляет 5 лет. Перерегистрация является своеобразной гарантией того, что уровень качества производства данного препарата становится, как правило, лучше, но никак не хуже. Переход фармпредприятий на систему GMP потребовал больших финансовых инвестиций. Действуют также специальные санитарные правила и методические указания по системе слежения. В каждой упаковке препарата имеется вкладыш с адресом предприятия-изготовителя и адресом национального органа контроля. Кроме того, действует ряд нормативно-правовых документов, регулирующих систему клинических испытаний GMP, ICH. Контроль выполнения утвержденных протоколов клинических испытаний лекарств осуществляют этические комитеты при медицинских учреждениях, а в лицензировании клинических баз активное участие принимает Минздрав России. Сама процедура проведения клинических исследований состоит из нескольких этапов (рис. 76).
Требует совершенствования система контроля деятельности учреждений, разрабатывающих новые фармпрепараты и проводящих клинические исследования. Разрабатываются принципы независимого аудита и государственной инспекции клинических испытаний ЛС. В настоящее время государственным контролем охватываются аналогичные ЛС и БАД. Это многокомпонентные смеси, состоящие из 20-40 ингредиентов растительного и животного происхождения, лекарственных трав, экстрактов и порошков всевозможных природных (и не только таковых) продуктов. Многие из них плохо изучены на предмет эффективности и безопасности использования, имеют противопоказания к применению, которые не отражены в инструкциях по их применению. По этой причине Управление по контролю за качеством пищевых продуктов и лекарственных средств (FDA) США выпускает многочисленные предостережения и рекомендации пациентам по прекращению приема тех или иных разновидностей БАД.
Основные мероприятия при назначении фармакологической экспертизы заключаются в следующем: • оценка качества и степени воздействия ЛС на организм больного; • обоснование рекомендаций по применению медикаментозной схемы лечения пациентов с определенной нозологией с учетом ряда признаков (половозрастных, профессиональных, предрасположенности к аллергическим реакциям и др.); • предупреждение возможного инициирования других (побочных) заболеваний. Те или иные осложнения фармакотерапии выявляются экспертами у 15-17% больных, а в 25% случаев фиксируется сомнительная целесообразность использования назначенных лекарств; • изучение и повышение качества лечения; • снижение его стоимости; • внедрение экономичных способов повышения гарантий доступной и высококвалифицированной медицинской помощи; • рационализация схем ресурсосберегающего потребления медикаментов. Подбор медикаментозного комплекса не всегда экономически правильный, особенно это касается такой сферы потребления ЛС, как стационарное лечение больных (так называемая полипрагмазия); • стимулирование познания врачами фармакологии современных ЛС; • усиление профессионального контроля использования ЛС и изделий медицинского назначения со стороны врачей, провизоров и фармацевтов; • проведение независимой оценки качества фармацевтической продукции, предлагаемой на рынке; • расширение перечня ассортимента фармацевтической и медицинской продукции, реализуемой через розничную сеть по единой лицензии организаций на фармацевтическую деятельность; • ускорение создания нормативно-методической базы лекарственного обеспечения граждан применительно к рыночным условиям на основе международных стандартов; • рациональность использования медикаментов; • ограничение агрессивной рекламы дорогостоящих ЛС; • защита прав застрахованных. В России на федеральном уровне действует единая контрольноразрешительная система в виде организационной структуры по контролю качества ЛС; на региональном уровне ее представляют юридически самостоятельные центры сертификации ЛС. С целью обеспечения населения и лечебных учреждений безопасными, эффективными и доступными ЛС и изделиями медицинского назначения в Российской Федерации действует система государственного контроля в сфере обращения медицинской продукции (рис. 77). Государственный контроль ЛС осуществляется в виде: • предварительного государственного контроля ЛС (впервые производимых, впервые ввозимых на территорию РФ, выпускаемых по измененной технологии или после перерыва в производстве от 3 лет и более, в связи с ухудшением качества); • выборочного государственного контроля ЛС (находящихся в обращении на территории РФ); • повторного выборочного государственного контроля ЛС (в случае возникновения споров об их качестве между субъектами обращения ЛС);
• инспекционного контроля (организаций-производителей, предприятий оптовой торговли ЛС, аптечных учреждений и учреждений здравоохранения). Сведения о регистрации содержатся в Государственном реестре ЛС, с которым можно ознакомиться в аптечных учреждениях. Все ЛС, реализуемые населению, должны иметь документы, подтверждающие соответствие их качества установленным требованиям. К таким документам относятся: • подлинник сертификата, выданного до 01.04.07, или декларации о соответствии; • копия сертификата, выданного до 01.04.07, заверенная держателем подлинника сертификата, нотариусом или органом, выдавшим сертификат; • товарно-сопроводительные документы, оформленные изготовителем или поставщиком, содержащие по каждому наименованию ЛС номер сертификата соответствия (выданного до 01.04.07), указания на срок его действия, орган, выдавший сертификат или регистрационный номер декларации о соответствии, срок ее действия, наименование изготовителя (продавца), принявшего декларацию, и орган, ее зарегистрировавший (эти документы должны быть заверены подписью и печатью изготовителя/поставщика с указанием его адреса и телефона). Важнейшей задачей системы фармаконадзора является мониторинг безопасности ЛС, уже обращающихся на рынке и широко применяемых в клинической практике. Его цель - выявление новых, не известных ранее нежелательных реакций и в случае их регистрации - своевременное внесение соответствующих изменений в действующие инструкции по медицинскому применению, а также информации об особенностях их развития, течения и лечения. Кроме того, задачами системы фармаконадзора являются распространение получаемых в ходе мониторинга данных через специализированные средства массовой информации, а также формирование и предоставление вниманию медицинской общественности объективной информации. В последние десятилетия повышается частота нежелательных реакций ЛС. К причинам развития этой негативной тенденции можно отнести: • широкое внедрение в клиническую практику новых препаратов с высокой биологической активностью; • полипрагмазию и нерациональное использование ЛС; • медицинские ошибки; • распространение некачественных и фальсифицированных ЛС; • растущую сенсибилизацию населения к биологически активным и химическим веществам; • взаимодействия с другими ЛС и продуктами питания; • фармакогенетические особенности пациентов; • в ряде случаев терапевтическую неэффективность в реальных клинических условиях; • ошибки в названиях ЛС. В настоящее время фармаконадзор, призванный давать всестороннюю оценку безопасности ЛС, стал уже самостоятельной медицинской дисциплиной. Фармаконадзор представляет собой направление науки и практической деятельности, связанной с выявлением, оценкой и профилактикой неблагоприятных последствий или любой другой проблемой, имеющей отношение к медицине. Таким образом, это понятие включает в себя не только мониторинг безопасности лекарств, но и любых других средств, применяемых в медицине. При этом ключевой проблемой оптимизации работы системы фармаконадзора, остро стоящей во всем мире, являются недостаточные объемы информации о нежелательных реакциях ЛС, направляемой специалистами здравоохранения в соответствующие регуляторные органы. ВОЗ укрепляет безопасность ЛС в рамках Международной программы мониторинга лекарств, которая начала действовать в 1968 г. Первоначально эта программа была пилотным проектом в 10 странах с установившейся национальной системой регистрации нежелательных реакций, но по мере того, как все большее число стран создавало национальные центры по контролю фармацевтической продукции для регистрации нежелательных реакций, эта сеть расширялась. В настоящее время в программе участвуют 86 стран. Главной задачей Программы мониторинга лекарств является, насколько возможно более раннее обнаружение «сигналов» о проблемах в области безопасности лекарств. Такой сигнал определяется ВОЗ как «поступившая информация о возможной причинной связи между неблагоприятным событием и лекарством», которая не была выявлена ранее. Информация о зарегистрированных случаях нежелательных реакций передается национальными центрами по контролю фармацевтической продукции в Сотрудничающий центр ВОЗ по международному мониторингу лекарств в Упсале (Швеция). Сообщения о таких случаях хранятся в базе данных нежелательных реакций ЛС. В ней насчитывается более 3,1 млн таких сообщений - это самый полный источник международной информации об осложнениях лекарственной терапии. Кроме того, ВОЗ также: • создала систему регулярного обмена информацией между государствами-членами о безопасности и эффективности фармацевтических препаратов с помощью сети специально назначенных национальных сотрудников по информации; • обеспечивает оперативную передачу национальным органам здравоохранения новой информации о серьезных нежелательных реакциях на фармацевтические препараты; • разрабатывает и распространяет руководящие принципы по созданию национальных центров мониторинга лекарств; • проводит среди врачей и специалистов по мониторингу безопасности лекарств во всем мире подготовку в области применения новых и комплексных ЛС (например, антиретровирусных препаратов); • проводит работу со странами по усилению органов, контролирующих лекарства, и созданию систем отчетности о нежелательных реакциях ЛС; • способствует налаживанию контактов между основными участниками (регулятивными органами, полицией, таможенными службами и т.п.) для борьбы с фальсифицированными лекарствами на национальном, региональном и глобальном уровнях. Будущее глобальной безопасности лекарств в значительной мере зависит от способности стран создать местные системы мониторинга лекарств, а также регистрации и хранения соответствующей информации. ВОЗ предполагает усилить свое техническое содействие национальным органам по контролю лекарств для достижения более полной согласованности в области проведения мониторинга лекарств и реагирования на сигналы о неблагоприятных реакциях лекарств на национальном и глобальном уровне. В России накоплен значительный опыт в области создания системы мониторинга безопасности ЛС. Так, еще в 1969 г. впервые был создан специальный Центр по изучению и регистрации побочного действия ЛС, который в 1973 г. был преобразован во Всесоюзный центр по изучению побочного действия ЛС. В 1997 г. открыт Федеральный центр по изучению нежелательных реакций ЛС, в составе которого была сформирована разветвленная сеть региональных центров. С 1999 г. и по сей день работа по мониторингу безопасности ЛС ведется сотрудниками Института доклинической и клинической экспертизы ЛС ФГУ «Научный центр экспертизы и средств медицинского применения». Региональные центры мониторинга безопасности ЛС продолжают активно функционировать на базах кафедр фармакологии вузов, ЛПУ, местных органов управления здравоохранением. Источниками получения информации о безопасности ЛС выступают сами компании-производители, врачи, провизоры и фармацевты, а в ряде случаев - и потребители фармацевтической продукции. Однако на сегодняшний день основным способом сбора информации о нежелательных реакциях ЛС в большинстве стран мира является метод спонтанных сообщений - добровольного предоставления субъектами сферы обращения ЛС данных о наблюдаемых ими нежелательных реакциях лекарств. К преимуществам использования метода спонтанных сообщений можно отнести его простоту, экономическую доступность, возможность контролировать безопасность препарата в течение всего времени его пребывания на рынке, охват большого количества пациентов; а к недостаткам - невозможность выявить весь спектр нежелательных реакций ЛС и установить их истинную частоту. Тем не менее постоянно ведется работа по совершенствованию процедуры предоставления спонтанных сообщений, направленная на увеличение объема получаемых данных. Так, в Российской Федерации разработаны специальные инструкции и унифицированные информационные карты, создана и пополняется централизованная база данных. Последствия нежелательных реакций ЛС нельзя недооценивать - зачастую они приводят к увеличению сроков стационарного лечения и последующей реабилитации, развитию осложнений, а в ряде случаев - и к летальному исходу. Во многих зарубежных исследованиях демонстрировалось, что прием лекарственных препаратов у 3-40% пациентов вызывал развитие лекарственных осложнений с серьезными (или фатальными) последствиями для здоровья. Было оценено, что в США смертность от нежелательных реакций лекарств занимает 4-6-е места. В исследованиях, проведенных в странах с отлаженной системой фармаконадзора, получены данные, согласно которым примерно 5% случаев госпитализации происходит вследствие нежелательных реакций лекарств, а 6-10% стационарных больных испытывают серьезные нежелательные реакции в период лечения в стационаре. Частота госпитализаций вследствие лекарственных осложнений достигает 11,5% в Норвегии, 13% - во Франции, 16% - в Великобритании. Однако эти цифры не отражают реального масштаба проблемы, поскольку в этих исследованиях исключены нежелательные события, вызванные другими, связанными с препаратом проблемами (например, передозировка, злоупотребление лекарственными травами, ненадлежащее и нерациональное применение лекарств, ошибки лечения и неэффективность лекарственной терапии). В последние годы по распространенности нежелательных реакций стабильно лидируют антибактериальные препараты, НПВП и средства, влияющие на сердечно-сосудистую систему. Однако следует отметить, что данное явление связано в основном не с истинной токсичностью препаратов данных групп, а, скорее, с частотой их использования. Более чем в половине случаев причиной возникновения нежелательных реакций является полипрагмазия - назначение нескольких ЛС одновременно, зачастую без учета их взаимодействия. Существенно повышает риск развития нежелательных реакций также нерациональное применение ЛС, в частности: недооценка анамнеза пациента, неправильный выбор ЛС и несоблюдение рекомендуемого режима их дозирования. Минимизировав указанные факторы, можно предупредить значительное количество лекарственных осложнений в реальной клинической практике, а это может быть достигнуто только путем информирования медицинской общественности об особенностях безопасности применения конкретных ЛС. Системный и более эффективный характер приобретает организация государственного контроля качества, эффективности и безопасности медицинской техники, эксплуатируемой в организациях здравоохранения. Разработана и действует программа мониторинга медицинской техники, ее сертификации и испытания изделий. Государственный контроль изделий медицинского назначения проводится: • на этапе производства: плановые (периодические) и внеплановые проверки организаций - производителей изделий медицинского назначения; • на этапе обращения. Контроль поступления в лечебные организации качественной медицинской техники обеспечивается через нормативное регулирование Минздрава РФ. Регулируется нормативная база государственной регистрации изделий медицинского назначения через взаимодействие организаций - элементов системы контроля: Комитета по новой медицинской технике Минздрава РФ и базовых испытательных центров страны. Испытание медицинской техники осуществляют более 130 учреждений здравоохранения, аккредитованных Минздравом РФ и уполномоченных на проведение клинических испытаний. Минздравом РФ ведется государственный реестр медицинской техники, в котором зарегистрировано около 2500 образцов отечественной медицинской техники и более 2 тыс. зарубежных образцов. Важным направлением государственного контроля является организация метрологического обеспечения средств измерения медицинского назначения. Во исполнение Федерального закона «Об обеспечении единства измерений» при органах управления здравоохранением в 55 субъектах РФ действуют базовые организации метрологической службы. Создано 3 региональных организационно-методических центра Минздрава РФ по техническому обслуживанию и метрологическому обеспечению учреждений здравоохранения. Формируется единый порядок проведения государственного метрологического контроля и надзора за средствами измерения медицинского назначения. Вопросы для повторения 1. Перечислите основные мероприятия по улучшению качества ЛС. 2. Назовите этапы и элементы проведения исследований фармацевтических препаратов. 3. В чем заключаются основные мероприятия к назначению фармакологической экспертизы? 4. Перечислите основные задачи государственного контроля в сфере обращения медицинской продукции. 5. Назовите виды государственного контроля ЛС. 6. Перечислите документы, подтверждающие качество ЛС. 7. Укажите цель мониторинга безопасности ЛС. 8. Назовите основные причины нежелательных реакций ЛС. 9. Назовите этапы государственного контроля изделий медицинского назначения. 10. Назовите основные элементы системы контроля безопасности медицинской техники.
|
|||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2017-01-19; просмотров: 733; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.128.190.102 (0.068 с.) |
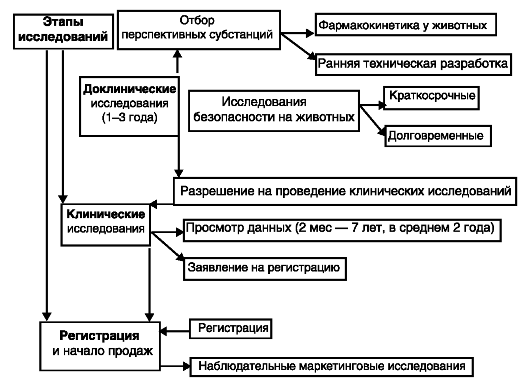 Рис. 76. Этапы проведения исследования фармацевтических препаратов
Рис. 76. Этапы проведения исследования фармацевтических препаратов Рис. 77. Задачи структуры контроля качества медицинской продукции
Рис. 77. Задачи структуры контроля качества медицинской продукции


