
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Основи хімічної термодинамікиСодержание книги Поиск на нашем сайте
Хімічна термодинаміка – розділ фізичної хімії, який вивчає енергетичні характеристики різних речовин (систем) і перетворення енергії в хімічних процесах. Термодинамічний (Т/д) метод дослідження є одним з ефективних засобів вивчення складних процесів з обміном масою і енергією в ізольованих, відкритих і закритих системах. Т\д система – тіло або сукупність тіл, що перебувають і взаємодії та відокремлені від довкола реальною або уявною поверхнею поділу. Можливий обмін енергією і речовиною з довкіллям. Тому розрізняють: 1) Ізольовані. Немає обміну ні енергією не речовинами. 2) закриті. Є обмін енергією і немає обміну речовинами. 3) Відкриті системи. Є обмін речовинами і енергією. Системи бувають: 1) Гетерогенні. Складаються з кількох фаз 2) Гомогенні. Складаються з однієї фази Фаза – це частина гетерогенної системи, відокремлена поверхнями поділу і має у будь-якому макроскопічному об’ємі однакові фізичні та хімічні властивості. Стан системи визначається сукупністю властивостей і характеризується параметрами певними величинами цих властивостей. Параметри – інтенсивні – залежать від розмірів системи (об’єм, маса, теплоємність) Та екстенсивні - властивості не залежать від розмірів системи (температура, тиск, концентрація, потенціал). Сукупність термодинамічних властивостей визначає термодинамічний стан системи. Якщо Т\д параметри стану з часом не змінюється, то система перебуває. У стані рівноваги, а стан рівноважний. Параметри у цьому разі рівноважні. Властивості системи, величини яких при переході з одного стану в інший залежить тільки від початкового та кінцевого стану системи і не залежить від шляху переходу, називають функціями стану. Розрізняють такі види процесів: Круговий (циклічний, коловий). Зміни функції стану по завершенню процесу = 0 Процеси є: 1) Оборотні. Які проходять у зворотному напрямку через ті самі проміжні стани без змін у системі та довкіллі. 2)Необоротні. Якщо при перебігу процесу в зворотному напрямку в системі або довкіллі відбуваються зміни Необоротні процеси перебігають довільно лише в одному напрямку – наближенні до рівноважного стану. Ізотермічний (T=const) Ізобаричний (P=const) Ізохорний (V=const) Адіабатичний (Q=0)
Мірою руху (основною властивістю матерії) є енергія. Це невідємна властивість системи. Розрізняють: кінетичну, або енергію руху і потенціальну, або енергію положення та взаємодії частинок системи. Повна енергія = кінетична + потенціальна як цілого + внутрішня енергія системи. Внутрішня енергія системи U – загальний запас енергії, що складається з кінетичної енергії руху її складових частин (молекул, атомів, іонів, електронів, тощо) та потенціальної енергії їхньої взаємодії без урахування кінетичної енергії системи та потенціальної енергії її положення. Величина U залежить від природи тіла (тіл), його (їхньої) маси, хімічного складу та параметрів, що зумовлюють стан системи P,V,T. Внутрішня енергія, віднесена до 1 моля як усі Т/д функції, є функцією системи. Її називають молярною внутрішньою енергією U (Дж/моль). Абсолютну величину U розрахувати неможливо. Але для термодинамічного аналізу системи достатньо знати лише приріст внутрішньої енергії ∆U або її зменшення. Приріст ∆U може бути додатнім або відємним. ∆U=UK-Un У термодинаміці поряд з внутрішньою енергією широко використовують поняття ентальпія H. Ентальпія – це енергія системи при сталому тиску. Чисельно вона дорівнює сумі внутрішньої енергії U та потенціальної енергії pV. H=U+pV Ентальпія, як і внутрішня енергія є функцією стану. Для хімічних та інших процесів, що звичайно перебігають при P=Const важливо знати не U, а H, оскільки остання враховує енергію, яка витрачається на зміну обє’му системи. Як і у випадку U, звичайно оперують величиною ∆H=HK+Hn
Передача енергії від системи до оточуючого середовища і навпаки відбувається у вигляді роботи A і теплоти Q. Робота (A)- це упорядкована форма передачі енергії. При передачі енергії у вигляді роботи система розвиває певним чином напрямлену силу, за рахунок якої виконується робота над іншою системою, до якої ця сила прикладена. Роботу, що виконує система над довкіллям умовимося вважати додатною A > 0 (+A), а роботу, що виконується над системою – від’ємного. A < 0 (-A) Передача енергії від однієї системи до іншої внаслідок неупорядкованого (хаотичного) руху молекул називають теплотою Q. +Q – додатна теплота – умовно називають кількість теплоти (ДЖ), яку система одержує від довкілля, а відє’мна теплота (-Q) – кількість теплоти, яку система віддає довкіллю.
Теплоємність – кількість теплоти, яка потрібна для нагрівання речовини на 1К. Теплоємність молярна – кількість теплоти, яку отримує 1моль речовини при збільшенні температури на 1 градус (в ДЖ/(моль. К)
При p=const
- ізобарна теплоємкість. Теплоємкість питома – для нагрівання 1гр. речовини (дж/ч.к) Середня теплоємність C= Частична теплоємкість при заданій температурі C= Відповідно При V= Теплоэмнысть при постыйному P і V, як H іU відрізняються на величену роботи, яка необхідна для зміни об’єму системи. Оскільки в процесі при p=const виконується робота, то для збільшення температури системи на одиницю треба більше теплоти. Ось чому Cp>Cv Cp=Cv+R Cp-Cv=R – це є робота ізобаричного розширення 1 моля ідеального газу при ∆T=1K Для рідин і твердих тіл Cp≈Cv 3.2 Закон Кіргофа Рівняння ……..-це рівняння Кіргофа в диференціальній формі а це в інтегральній формі Отже, можна визначити теплові ефекти в залежності від температури.
1) T.T - ∆С > 0 зі збільшенням температури тепловий ефект реакції зростає. 2) При ∆С ˂ 0 - ∆Н зменшується. Ізобарна та ізохорна теплоємності відрізняються (як і Н та U) на величину роботи розширення. Оскільки в процесі при p=const виконується робота, то Cp>Cv: Cp = Cv + R Тепловий ефект хімічних реакцій залежить від температури:
Це також математичний запис закону Кірхгофа. Температурний коефіцієнт теплового ефекту дорівнює різниці між теплоємкостями вихідних речовин і продуктів реакції.
3.3 Основні принципи (початки) термоденаміки. 3.3.1 Перший та другий початки термодинаміки. Ізотермічний процес. Адіабатичний процес. Перший закон термодинаміки є законом збереження енергії в застосуванні до термічних явищ. Якщо система бере участь в тому чи іншому процесі, то підведена до неї теплота може збільшувати внутрішню енергію системи на ∆U і використовується на завершення роботи А, тобто q=∆U + A Це рівняння є аналогічним вираженням першого закону термодинаміки. Для замкненої (ізольованої) системи q = 0 і А = 0,, відповідно ∆U = 0, що і потрібно за законом збереження енергії в ізольованій системі. Вираз Q = ∆U + A Означає: «При нагріванні будь-якої системи теплота витрачається на виконання роботи на збільшення внутрішньої енергії», або «В ізольованій системі сума усіх видів енергії стала». Це і є формулюваннями першого початку (закону) термодинаміки. Зміна ∆U не залежить від шляху процесу, але робота А, як було вказано на прикладі розширення ідеального газу, залежить від шляху процесв; відповідно, і Q також залежить від шляху процесу. Тому робота А і теплота Q не є функціями стану системи. При постійному об’ємі робота розширення А = 0; якщо, крім цього, ніяких інших робіт не відбувається (наприклад, електричною), то вся теплота, яка підведена до системи, використовують (витрачається) тільки на збільшення її внутрішньої енергії: qv = ∆U При постійному тиску відведена енергія в вигляді теплоти qp йде на збільшення внутрішньої енергії і здійснення роботи розширення: qp = ∆U +p(V2-V1), або qp=U2-U1+pV2-pV1=(U2+pV2)-(U1-pV1) Із останнього рівняння можна зробити висновок про те, що енергія, яка підведена до системи в вигляді теплоти при постійному тиску, витрачається на прирощення деякої функції стану:
H = U + pV Ця функція називається ентальпією. Її зміни, як і зміни внутрішньої енергії, не залежить від шляху процеса, оскільки зміни об’єму при постійному тиску визначається тільки початковими і кінцевим станом системи. qp = H2 - H1 = ∆H. Якщо система якимось чином повертається назад (круговий процес), то зміна будь – якого параметру дорівнює нулю, а звідки ∆U = 0 і ∆H = 0. II Початок термоденаміки. Швидко чи повільно (це рівноважна термоденаміка не розглядає), але всяка система направлена до стану істинної рівноваги. Це може служити одним із формулювань другого початку термоденаміки. Його формулюванням можна прийняти і неможливість самовільного переходу теплоти від менш нагрітого тіла до більш нагрітого. Також, згідно другого закону, якщо періодично діюча машина збирає від нагрівача який має температуру ТН, теплоту Q і перетворює її на роботу А, то завжди А ˂ Q і різниця Q-А переходить в вигляді теплоти в холодильнику з температурою Тх (вигляді компенсації за роботу). Робота може досягнути максимального значення: Аmax, a(Q-Amax) – мынымального при зворотному ведены процессу і при використанні ідеальної машини (без тертя); тоді відношення В реальних процесах,які майже ніколи не бувають оборотними, відношення Якщо в системі проходить реакція при постійному тиску і температурі, то зміни ентальпії також може бути запропоновано в вигляді двох доданків: зміни вільної енергії ∆G і зміни зв’язаної енергії ∆L. Першу частину енергії принципово можна перетворити на корисну роботу Аmax при оборотному ізобарно – ізотермічному веденні процесу, а другу – ні. Можна записати.
Де ∆G є зміна вільної енергії Гіббса при постійному тиску і температурі, її називають змінами ізобарно – ізотермічного потенціалу. Зв’язана енергія ∆L виражається добутком абсолютної температури Т на зміни функції стану, яка називається ентальпієюS. ∆L = T∆S. Тоді ∆G = Ізометричні процеси здійснюються при постійному Т, ізобаричний – при постійному тиску (Р), ізохорний - при постійному об’ємі (V).
Процеси, в яких може здійснюватися робота, але система не обмінюється теплотою з навколишнім середовищем, називається адіабатичними. Перед тим, як узагальнювати відомості про початки (начала) термодинаміки нагадаємо: Коефіцієнт корисної дії ККД (рос.«КПД»):
де
Власне, це є математичним виразом ІІ початку термодинаміки, яке можна формулювати так: «Тепло не може довільно переходити від холодного (менш теплого) до нагрітого (теплішого) тіла» (визначення Р.Клазузіуса). Серед інших визначень ІІ початку термодинаміки є таке: «Неможливо створити машину, яка б перетворювала усе тепло у роботу», а також «Неможливо створити вічний двигун». Суть ІІ початку термодинаміки випливає з того, що малоймовірно щоб хаотичний тепловий рух молекул перейшов у напрямлений рух. Навпаки, напрямлений рух молекул може повнісю перейти у хаотичний (робота може повністю перейти в теплоту). Хаотичний рух молекул є природнім. Це є причиною того, що різні види енергії прагнуть у теплоту, а теплота передається менш нагрітим тілам. 3.3.3. Ентропія. Отже довільні процеси відбуваються з розсіюванням теплової енергії. Для кількісної характеристики цього процесу Рудольф Клаузіус (1865р.) увів термодинамічну функцію S і назвав її енергією:
Знак нерівності – для необоротних процесів а = - для оборотних. Одиниця виміру. Ентропія – є мірою розсіяної (знеціненої) енергії. Чим більше енергія, тим менша частина енергії може перетворитися в роботу. Отєе, енергія – міра необоротності процесу. Довільно відбувається процеси, в яких ентропія зростає. Це також може бути формуванням ІІ початку термодинаміки dS>0 δQ = TdS Отже TdS ≥ dU + pdv
Це є об’єднаний вираз ІІ та І початків термодинаміки.
Австрійський фізик Людвіг Больцман показав, що ІІ початок термодинаміки має статистичний характер і свідчить лише про ймовірність процесу. Больцман встановив зв'язок між ентальпією S та ймовірністю стану системі W (від німецького W ahrscheinlichkeit.) В англомовній заміст W використовують P від англійського P robability). S = k · ln W k – стала Больцмана (k = Чим більшою кількістю мікрочастинок представлена система, тим більше варіантів розподілу цих частинок, тим вище значення ентропії і тим більш знаціненим буде вмщений у системі запас енергії. Отже, ентропія характеризує відносну цінність енергії, яку має система, тобто характеризує ту частину енергії, яка не перетворюється в роботу.
ІІІ Початок термодинаміки. ІІІ Початок термодинаміки. S T=0 дорівнює 0; lim ST→0 = 0 Дійсно, при T=0 W=1 S = k · lnl = 0
Термодинамічні потенціали Ентропія є функцією, що визначає можливість перебігу довільного процесу в ізольованій системі. Для закритих систем функціями, які визначають таку можливість є термоденомічні потенціали: Ізохорично – ізотермічний потенціал, або вільна енергія Гельмгольца (А) та ізобарично – ізотермічним потенціал, або енергія Гіббса. У більшості випалків (в хімії та поліграфічних технологіях) енергія Гіббса частіше застосовується, оскільки процеси проходять при сталому тиску, а не при сталому обємі.
A=U-TS (∆A=∆U - T∆S) G=H- TS (∆G=∆H - T∆S)
Абсолютні значення Т/д потенціалів. Не відомі. Звичайно користуються величини ∆A Та ∆G, КД (моль). Зв'язок А та G: G = U + pV – TS = A + pV -∆G=-∆A-pV
Зменшення енергії гіббса більше або дорівнює максимальній корисній роботі процесу. G тa А називають також вільною енергією. Т/д потенціали не змінюються в оборотних процесах і зменшуються – в необоротних.
|
|||||||||
|
|
Последнее изменение этой страницы: 2016-09-19; просмотров: 561; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 13.58.3.190 (0.009 с.) |


 , вірніше Cp= (
, вірніше Cp= ( ,
,

 –ізохорна теплоємність.
–ізохорна теплоємність.

 , яка має нахву коефіцієнтом корисної дії, такоє досягае максимальне значення, яке дорівнює:
, яка має нахву коефіцієнтом корисної дії, такоє досягае максимальне значення, яке дорівнює: 
 завжди менше відношенню
завжди менше відношенню  . Це означає, що в найкращому випадку частину забраної від нагрівача енергії в вигляді теплоти може бути використана як корисна частина в вигляді роботи Аmax. Ця величина характеризує так звану вільну енергію, а інша частина ні за яких умов не може бути перетворена на роботу – ця зв’язана енергія.
. Це означає, що в найкращому випадку частину забраної від нагрівача енергії в вигляді теплоти може бути використана як корисна частина в вигляді роботи Аmax. Ця величина характеризує так звану вільну енергію, а інша частина ні за яких умов не може бути перетворена на роботу – ця зв’язана енергія.
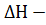 T∆S і G = Н - TS.
T∆S і G = Н - TS.
 ,
, - температура нагрівника.
- температура нагрівника.  - температура холодильника.
- температура холодильника.
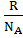 )
)


