
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Гемолитические анемии, таким образом, регенераторные или гиперрегенераторные, чаще всего – умеренно гипохромные, нормобластические.Содержание книги
Поиск на нашем сайте Общая клиническая характеристика гемолитических анемий. Острый внутрисосудистый гемолиз – картина шока с характерными гемодинамическими нарушениями вследствие активации при цитолизе сторожевой полисистемы плазмы, резкое повышение температуры, ДВС синдром, гемоглобинурия, гиперкалиемия, приводящая к развитию аритмий. Хронические гемолитические анемии, для них характерны гепато-спленомегалия, гемосидероз, холелитиаз, возможна иктеричность склер, желтуха, накопление железа в организме (гемосидероз), кризовое течение. Кризы: - гемолитический (вне криза нормальное состояние крови), - апластический (обусловлен парвовирусом В19), - мегалобластический (дефицит фолиевой кислоты, результат усиленного потребления вследствие гиперактивного эритропоэза), - окклюзионный (аггрегация серповидных эритроцитов при серповидноклеточной анемии), - секвестрационный (внезапное переполнение селезенки кровью, снижение ОЦК); Классификация гемолитических анемий Гемолитические анемии делятся на две основные группы – наследственные и приобретенные. Наследственные гемолитические анемии Мембранопатии – анемии, вызванные наследуемыми нарушениями структуры мембраны эритроцитов: а) белковозависимые мембранопатии: микросфероцитоз (болезнь Минковского - Шоффара), эллиптоцитоз, стоматоцитоз; б) липидозависимые мембранопатии (акантоцитоз). Ферментопатии – нарушения активности ферментов, обеспечивающих жизнеспособность эритроцитов: пентозофосфатного цикла, гликолиза, глютатиона, участвующих в использовании АТФ, рибофосфатпирофосфаткиназы. Наиболее частые: а) дефицит глюкозо-6-фосфатдегидрогеназы (Г-6-ФДГ); б) дефицит пируваткиназы. Гемоглобинопатии – нарушения структуры гемоглобина а) нарушения первичной структуры цепей глобина (гемоглобинозы): серповидно-клеточная анемия (гемоглобиноз S), гемоглобиноз С; б) замедление или отсутствие синтеза одной из цепей глобина (талассемии): альфа-талассемия, бета-талассемия. Мембранопатии. Наследственный микросфероцитоз (болезнь Минковского – Шоффара) распространен в Скандинавии, Балтике (частота 1:5000). Тип наследования – аутосомно-доминантный. В основе заболевания лежит дефект генов, кодирующих синтез подмембранных белков цитоскелета эритроцитов, чаще спектрина и анкирина. Эритроциты приобретают шаровидную форму, теряют способность к деформации, повреждаются в капиллярах. В селезенке они частично разрушаются макрофагами, превращаясь в микросфероциты. Кроме того, дефектная мембрана эритроцитов становится высокопроницаемой для ионов натрия и воды, удаление избытка которых требует больших энергетических затрат, что также сокращает срок их жизни до 12-14 дней. В мазке крови такие эритроциты не имеют центрального просветления.
Для сфероцитов характерна сниженная осмотическая стойкость, цветовой показатель = 1, кризовое течение; вне кризов анемию, как правило, не обнаруживают. Количество ретикулоцитов увеличено в период ремиссии и особенно после гемолитического криза - 10-15 и 50-60% соответственно. При тяжелых кризах удаление селезенки дает хороший эффект. Дефекты белков мембраны эритроцитов могут приводить также к редким формам гемолитической анемии - эллиптоцитозу, стоматоцитозу. Другой вид мембранопатии - аномальные липиды в составе мембраны эритроцитов, в них появляются выпячивания мембраны и они становятся похожими на листья растения аканта, поэтому их называют акантоцитами (другое название - шпоровидные клетки).
Низкая деформируемость, пониженная резистентность таких эритроцитов к различным воздействиям являются причиной их повышенного распада и развития анемии. Подобные изменения эритроцитов встречаются, в частности, у больных наследственной абеталипопротеинемией, при циррозе печени, при нарушении липопротеинового обмена, при авитаминозе Е, у лиц с удаленной селезенкой. Ферментопатии В настоящее время известно более 20 наследственных энзимопатий эритроцитов, которые приводят к повышенному гемолизу. К ним относится нарушение активности ферментов гликолиза, пентозофосфатного цикла, системы глютатиона, метаболизма адениннуклеотидов и др. Наиболее частая ферментопатия – дефицит глюкозо-6-фосфат-дегидрогеназы (Г-6-ФДГ), он отмечается в районах распространения малярии. Считается, что приблизительно 1/20 человечества имеет дефект фермента Г-6-ФДГ. Гемолитическая анемия, обусловленная недостаточностью Г-6-ФДГ Известно большое количество мутантных форм Г-6-ФДГ. (от 90 до 250 по данным разных авторов), из которых две являются основными: более легкая африканская форма дефицита Г-6-ФДГ - тип А и более тяжелая средиземноморская - тип В, при которой снижена активность и количество фермента. Наследование дефицита Г-6-ФДГ сцеплено с X-хромосомой, поэтому среди заболевших лиц преобладают мужчины. При недостаточной активности Г-6-ФДГ нарушается пентозофосфатный цикл в эритроцитах, в результате нарушается восстановление НАДФ и восстановление глютатиона. Восстановленный глютатион защищает гемоглобин и мембрану эритроцитов от различного рода окислителей, образующихся при инфекциях, приеме определенных лекарственных препаратов (антималярийных, противотуберкулезных, сульфаниламидов, нитрофуранов, анальгетиков и пр.), при употреблении в пищу бобов vicia vafa. В результате действия окислителей в эритроцитах происходит преципитация гемоглобина и отложение его в виде телец Гейнца, ускоренное старение эритроцитов.
Повышается проницаемость мембраны для натрия и воды, что способствует повреждению. Гемоглобинопатии Гемоглобинопатии преимущественно поражают население тропических и субтропических областей (Экваториальная Африка, Аравийский полуостров, Южная Индия, Южный Китай, Средиземноморье, Азербайджан, Грузия).Наиболеераспространены и отличаются тяжестью проявлений серповидноклеточная анемия и большая талассемия, или анемия Кули. Большинство гемоглобинопатий клинически не проявляются; при некоторых - могут наблюдаться: анемия, эритроцитоз или цианоз (например, при метгемоглобинопатиях). а) Нарушения первичной структуры глобиновых цепей гемоглобина вследствие генной мутации. Описано около 500 аномальных гемоглобинов. Серповидно-клеточная анемия (СКА) Тип наследования аутосомно-рецессивный, дефект бета-глобиновых цепей, в которых в 6 положении гидрофильный глютамин заменен гидрофобным валином, образуется HbS. Восстановленная форма HbS мало растворима. При гипоксемии и снижении скорости кровотока HbS полимеризуется в длинные нерастворимые нити, растягивающие эритроциты в форме серпа. Если содержание HbS больше 45% (гомозиготное состояние), образуются эритроциты необратимо серповидной формы, склонные к агрегации, что повышает вязкость крови, вызывает закупорку сосудов (вазоокклюзию), нарушение микроциркуляции и боль.
В этом случае точечная мутация обусловливает нарушение структуры гемоглобина, гемолиз, анемию, а также закупорку сосудов и нарушение кровообращения.
Больные СКА имеют типичный вид: удлиненный нижний сегмент тела, выступающий лоб, «башенный» череп, гепато-спленомегалия. Самый характерный криз для этого заболевания - вазоокклюзионный, проявляющийся резкой болью. Окклюзия сосудов может развиваться в разных органах, поэтому клинические симптомы чрезвычайно разнообразны. В мазках крови можно обнаружить серповидные клетки.
Примерно одна треть обитателей тропических и субтропических регионов Африки (т. наз. «малярийного пояса») являются носителями признака серповидноклеточности. Они не страдают от гемолитической анемии, в то же время более устойчивы по отношению к малярии. Талассемии - замедление или отсутствие синтеза одной из цепей глобина: α-талассемия, ß-талассемия. При талассемиях мутации располагаются не в структурных генах, а в генах-регуляторах, поэтому структурных нарушений нет, а результатом мутаций служит замедление или отсутствие синтеза одной из глобиновых цепей и замена ее синтезом другой цепи. Талассемия встречается в странах Средиземноморья, в Китае, Индии, в Европе, у жителей Закавказья и Средней Азии. Самая высокая заболеваемость – на Мальдивах, где носители признака составляют 18%. Гетерозиготная бета-талассемия наблюдается у 7— 10 % населения в низменных районах Азербайджана. Альфа-талассемия – полное или частичное прекращение синтеза α-цепей. Компенсаторно синтезируются: а) в пренатальный период γ-цепи - образуется тетрамер γ (Hb Барт); б) в постнатальный – тетрамер ß (HbH). Синтез α-цепей кодируют 4 гена, поэтому степень нарушения их синтеза меньше, чем при ß-талассемии; выраженный дисбаланс развивается только тогда, когда поражены все 4 гена. Агрегаты из ß-цепей более растворимы, чем агрегаты из α -цепей, поэтому гемолиз при α-талассемии выражен слабее, чем при ß-талассемии, а эритропоэз более эффективен. Бета-талассемия обусловлена снижением скорости синтеза ß-цепей гемоглобина (ß+-талассемия) или отсутствием их синтеза (ß0-талассемия). Неповрежденные α-цепи избыточно накапливаются в клетках, что ведет к повреждению мембраны и разрушению эритроидных клеток в костном мозге (неэффективный эритропоэз) и эритроцитов в крови. Деструкция эритроидных клеток способствует гиперплазии костного мозга, что отражается на структуре скелета, ведет к повышенному всасыванию железа и перегрузке организма железом. Тяжелая гомозиготная форма ß-талассемии - болезнь Кули, или большая талассемия. Кроме того, выделяют промежуточную, малую и минимальную талассемию.
значительная спленомегалия,
желтушность, хронические язвы на нижних конечностях, башенный череп,
осмотическую стойкость вследствие избытка поверхности мембраны.
Сканирующая электронограмма. Стрелками показаны два кодоцита («хвостатые клетки») – это другое название мишеневидных клеток.
|
||
|
|
Последнее изменение этой страницы: 2016-08-12; просмотров: 199; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.113 (0.011 с.) |






 http://theblackbottom.com/?attachment_id=8214
http://theblackbottom.com/?attachment_id=8214

 уплощенная переносица. Скулы выступают, глазные щели сужены, нарушены прикус и расположение зубов.
уплощенная переносица. Скулы выступают, глазные щели сужены, нарушены прикус и расположение зубов.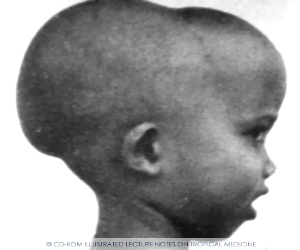

 В мазках крови эритроциты мишеневидные, имеют повышенную
В мазках крови эритроциты мишеневидные, имеют повышенную



