
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Условные обозначения и запись хромосомного дисбаланса по международной цитогенетической номенклатуре.Содержание книги
Поиск на нашем сайте
Для обозначения структурных нарушений хромосом Международная система цитогенетической номенклатуры хромосом человека рекомендует следующие символы и сокращения. а) Краткий перечень символов и сокращений, принятых в цитогенетике: асе - ацентрический фрагмент: add - дополнительный материал неизвестного происхождения; сеn - центромера; chi - химеризм; chr - хромосома; del -делеция: der - дериват, т.е. источник происхождения вновь образованной хромосомы; dic - дицентрическая хромосома; dup - дупликация; fem - женский пол; fis - центромерный разрыв; fra - ломкий участок; g - пробел; h -гетерохроматин; i - изохромосома; idic - изодицентрическая хромосома; ins - вставка; inv - инверсия; q -длинное плечо хромосомы; mal - мужской пол; mar - маркерная хромосома неизвестного происхождения; mat - материнского происхождения; mos - мозаик; р - короткое плечо хромосомы; pat -отцовского происхождения; г - кольцевая хромосома; гcр - реципрокная перестройка; rob - робертсоновская транслокация; s - спутник; sce - сестринские хроматидные обмены; t - транслокация; tan - тандемная перестройка; tel - теломера; ter - терминальный конец хромосомы; upd - униродительская (однородительская) дисомия. б) Обозначение числовых нарушений хромосом. При описании кариотипа сначала указывается общее число хромосом, включая половые, а затем через запятую без пробела - набор половых хромосом. Таким образом, нормальный кариотип человека обозначается следующим образом: 46,XX - нормальный женский и 46,XY - нормальный мужской кариотип. Изменение в системе половых хромосом отмечается путем непосредственного их перечисления после указания общего числа хромосом в наборе. Лишняя или недостающая аутосома при конституциональных или приобретенных хромосомных нарушениях обозначается знаком "+" или "-", который ставится перед номером хромосомы через запятую после перечисления половых хромосом: 45,Х - кариотип с одной X хромосомой (синдром. Шерешевского-Тернера); 47,XXY - кариотип с двумя X хромосомами и одной Y хромосомой (синдром Клайнфельтера); 47,XYY - кариотип с одной X идвумя Y хромосомами; 47,ХХХ - кариотип с тремя X хромосомами; 47,ХХ,+21 - кариотип с трисомией по 21-й хромосоме (синдром Дауна); 47,ХХ,+13 - кариотип с трисомией по 13-й хромосоме (синдром Патау); 47,ХХ,+18 - кариотип с трисомией по 18-й хромосоме (синдром Эдвардса-Смита);
46,ХХ,+8,-21 - кариотип с трисомией 8 и моносомией 21. Для отличия между мозаицизмом (наличия в одном организме нескольких клеточных линий, возникших из одной зиготы) и химернзмом (наличия в одном организме нескольких клеточных линий, возникших из разных зигот) используются сокращения mos и chi - mos 45,X/46,XX и chi 46,XX/46,XY соответственно. В случае мозаицизма нормальный диплоидный клон клеток, если он присутствует, всегда указывается последним - mos 47,XXY/46,XX. Если имеется несколько аномальных клеточных клонов, то сначала указывается наибольший, а остальные перечисляются по мере убывания. При этом независимо от доли нормального клеточного клона он всегда указывается последним. В квадратных скобках указывается число обнаруженных клеток с данным кариотипом - mos 45,Х[15]/47,ХХХ[10]/46,ХХ[23]. Наибольший из имеющихся клеточных клонов у химер указывается первым: chi 46,XX[25]/46,XY[10]. в) Обозначение структурных перестроек хромосом. При наличии структурных хромосомных нарушений для описания кариотипа применяется как короткая форма записи аномального кариотипа, указывающая только на характер хромосомного нарушения, так и развернутаяформа, обозначающая конкретные поврежденные сегменты хромосом и степень протяжения вовлеченных в перестройку фрагментов хромосом. Одно двоеточие (:) указывает на разрыв хромосомы, а двойное двоеточие (::) указывает на разрыв и воссоединение. Для избегания громоздкой записи хромосомного дисбаланса между точками разрывов хромосомы часто указывают стрелку (—>), означающую протяженность фрагмента хромосомы от одного участка до другого. При наличии повреждения структуры одной хромосомы, после символа обозначающего характер этого повреждения, в круглых скобках указывается номер этой хромосомы. Если известны точки повреждения хромосомы, то они перечисляются в других круглых скобках с указанием плеча, региона и сегмента аберрантной хромосомы. Символ del используется для обозначения как терминальных, так и интерстициальных делеций. Стрелка (—>) не используется в короткой системе записи. 46,XX,dup(5)(pl5) - дупликация сегмента р15 в 5 хромосоме; 46,XX,del(5)(ql3) - терминальная делеция длинного плеча 5 хромосомы в сегменте ql3 (короткая форма записи) или 46,XX,del(5)(pter—>ql3:) - терминальная делеция длинного плеча 5 хромосомы в сегменте ql3. Оставшаяся в результате делеций хромосома 5 состоит из целого короткого плеча - от его терминального участка и части длинного плеча – до сегмента 5ql3 с разрывом в этом сегменте (развернутая форма записи);
При перестройке двух хромосом с двумя разрывами, сначала в круглых скобках указываются номера этих хромосом, между которыми ставится знак ";", а затем в других круглых скобках соответственно указываются поврежденные плечи этих хромосом и точки разрывов. Первой указывается хромосома с более низким номером. Однако, если в перестройке участвует половая хромосома, то она указывается первой: 46,XУ,t(12;16)(q13;p11) - транслокация между хромосомами 12 и 16 с двумя точками разрывов, один из которых затрагивает сегмент 13 длинного плеча хромосомы 12, а второй - сегмент 11 короткого плеча хромосомы 16. Упомянутое выше правило первого указания хромосомы с более низким номером или половой хромосомы не применяется в случае перестройки хромосом с тремя разрывами, когда часть одной хромосомы вставлена в другую. Если какой-либо участок хромосомы встраивается в другой сегмент той же самой хромосомы, то точка встраивания всегда указывается первой. В случае прямой ннсерции оставшиеся точки поломки указываются следующим путем: сначала более проксимальная точка, а затем более дистальная. В случае инвертированной ннсерции применяется обратная запись - сначала указывается дистальная точка разрыва, а затем проксимальная: 46,XX,ins(2)(ql3pl3p23) - прямая вставка фрагмента короткого плеча хромосомы 2 (участка р!3-р23) в сегмент q13 этой же хромосомы. Дериваты хромосом или структурно перестроенные хромосомы, обозначаемые символом der, образуются в результате: 1) двух и более перестроек в пределах одной хромосомы, например инверсии и делеции той же самой хромосомы или делеции обоих плеч одной хромосомы; 2) перестройки, вовлекающей две и более хромосомы, например насбалансированный продукт транслокации. Термин "дериват" всегда применяется к хромосоме, имеющей интактную центромеру. Перестроенная хромосома указывается в скобках за которыми перечисляются все аберрации, вовлеченные в образование этого хромосомного деривата: 46,XX,der(9)del(9)(pl2)del(9)(q31) или 46,XX,der(9)(:pl2->q31:) - дериват хромосомы 9, возникший в результате терминальных делеции в коротком и длинных плечах с точками поломок в сегментах 9р12 и 9q31; Робертсоновские транслокации, возникающие путем центрического слияния длинных плеч акроцентрических хромосом 13-15 и 21-22 с потерей их коротких плеч могут быть адекватно описаны с использованием символа der или rob: 45,XX,der(13;21)(13qter-+13ql0::21ql0—>21qter) - развернутая форма записи робертсоновской транслокации между 13 и 21 хромосомами; 46,XX,der(21;21)(q10;q10),+21 - транслокационный вариант синдрома Дауна; Транслокации целых плеч хромосом описываются обозначением точек разрывов в центромерных районах р10 и q10. При сбалансированных обменах целыми плечами двух разных хромосом точка разрыва в хромосоме более низкого номера или в половой хромосоме указывается как р10: 46,XY,t(l;3)(p10;q10) - краткая форма записи реципрокной транслокации целых плеч хромосом 1 и 3. Короткое плечо хромосомы 1 слилось в центромерной области с длинным плечом хромосомы 3, а длинное плечо хромосомы 1 - с коротким плечом хромосомы 3.
Термин "филадельфийская хромосома" сохранился по историческим причинам для описания деривата хромосомы 22, образованного в результате транслокации t(9;22)(q34;q11) при хроническом миелолейкозе. Аббревиатура Ph может быть использована в тексте, но не для описания кариотипа, который указывается как 46,XУ,der(22)t(9;22)(q34;q11). Символ diс используется для описания дицентрических хромосом, а idic - для изодицентрических хромосом. Дицентрическая хромосома рассматривается как одна хромосома: 45,XY,dic(13;15)(q22;q24) - дицентрическая хромосома, образованная хромосомами 13 и 15, замещающая эти две нормальные хромосомы (краткая форма записи); Символ i применяется для обозначения изохромосом, образующихся в результате разрыва в центромерном районе хромосом в сегментах р10 и q10 в соответствии с морфологией изохромосомы: 46,X,i(X)(qlO) или 46,X,i(X)(qter—>qlO::qlO—>qter) - кариотип с одной нормальной X хромосомой и изохромосомой X по длинному плечу; Аномальная хромосома, в которой ни одна из ее частей не может быть однозначно идентифицирована с помощью традиционных методов дифференциального окрашивания хромосом обозначается как маркерная (mаг) хромосома: 47,ХХ,+таг - кариотип с одной маркерной хромосомой. Фрагильные или ломкие сайты хромосом, обозначаемые символом fra, могут встречаться как нормальные варианты хромосом или могут быть связаны с аномалиями фенотипа. Они проявляют себя как участки хромосом, обнаруживающие разрывы или пробелы хроматид, в ответ на определенные условия культивирования клеток или действие специфических реагентов. Выделяют 2 типа фрагильных сайтов - наследуемые (около 30) и обычные или конститутивные (около 80). Из всех фрагильных сайтов хромосом важное клиническое значение имеет ломкий сайт Xq27.3, экспрессирующийся у некоторых индивидов в условиях культивирования клеток с низким содержанием в питательной среде фолиевой кислоты. У лиц с этим наследуемым фрагильным сайтом (обозначаемым FRAXA) выявляется один из наиболее частых синдромов умственной отсталости - синдром фрагильной или ломкой Х-хромосомы или синдром Мартина-Белл: 46,X,fra(X)(q27.3) - ломкая хромосома X в локусе q27.3 у женщины; 46,Y,fra(X)(q27.3) - ломкая хромосома X в локусе q27.3 у мужчины. Хромосомные болезни а) Классификация хромосомных болезней. Хромосомные болезни - это большая группа врожденных наследственных болезней. Они занимают одно из ведущих мест в структуре наследственной патологии человека. По данным цитогенетических исследований среди новорожденных детей частота хромосомной патологии составляет 0,6— 1,0%. Самая высокая частота хромосомной патологии (до 70%) зафиксирована в материале ранних спонтанных абортусов. Следовательно, большинство хромосомных аномалий у человека несовместимо даже с ранними этапами эмбриогенеза. Такие зародыши элиминируются во время имплантации (7-14-е дни развития), что клинически проявляется как задержка или выпадение менструального цикла. Некоторая часть эмбрионов гибнет вскоре после имплантации (ранние выкидыши). Сравнительно немногие варианты числовых аномалий хромосом совместимы с постнатальным развитием и ведут к хромосомным заболеваниям (Кулешов Н.П., 1979).
Хромосомные болезни появляются вследствие повреждений генома, возникающих при созревании гамет, в процессе оплодотворения или на ранних стадиях дробления зиготы. Все хромосомные болезни могут быть разделены на три большие группы: 1) связанные с нарушением плоидности; 2) обусловленные нарушением числа хромосом; 3) связанные с изменением структуры хромосом. Аномалии хромосом, связанные с нарушением плоидности, представлены триплоидией и тетраплоидией, которые встречаются преимущественно в материале спонтанных абортусов. Отмечены лишь единичные случаи рождения детей-триплоидов с тяжелыми пороками развития, несовместимыми с нормальной жизнедеятельностью. Триплоидия может возникать как вследствие дигении (оплодотворение диплоидной яйцеклетки гаплоидным сперматозоидом), так и вследствие диандрии (обратный вариант) и диспермии (оплодотворение гаплоидной яйцеклетки двумя сперматозоидами). Хромосомные болезни, связанные с нарушением числа отдельных.хромосом в наборе, представлены либо целой моносомией (одной из двух гомологичных хромосом в норме) либо целой трисомией (тремя гомологами). Целая моносомия у живорожденных встречаются только по хромосоме X (синдром Шерешевского-Тернера), поскольку большинство моносомий по остальным хромосомам набора (Y хромосоме и аутосомам) погибают на очень ранних этапах внутриутробного развития и достаточно редко встречаются даже в материале спонтанно абортированных эмбрионов и плодов. Следует, однако, отметить, что и моносомия X с достаточно высокой частотой (около 20%) выявляется у спонтанных абортусов, что свидетельствует о ее высокой пренатальной летальности, составляющей свыше 99%. Причина гибели зародышей с моносомией X в одном случае и живорождения девочек с синдромом Шерешевского-Тернера в другом, неизвестна. Существуют ряд гипотез, объясняющих этот факт, одна из которых связывает повышенную гибель Х-моносомных зародышей с более высокой вероятностью проявления рецессивных летальных генов на единственной Х-хромосоме. Целые трисомии у живорожденных встречаются по X, 8, 9, 13, 14, 18, 21 и 22 хромосомам. Наибольшая частота хромосомных нарушений - до 70% отмечается у ранних абортусов. Трисомии по 1, 5, 6, 11 и 19 хромосомам встречаются редко даже в абортивном материале, что свидетельствует о большой морфогенетической значимости этих хромосом. Более часто целые моно- и трисомии по ряду хромосом набора встречаются в мозаичном состоянии как у спонтанных абортусов, так и у детей с МВПР (множественными врожденными пороками развития).
Хромосомные болезни, связанные с нарушением структуры хромосом, представляют большую группу синдромов частичных моно- или трисомии. Как правило, они возникают в результате структурных перестроек хромосом, имеющихся в половых клетках родителей, которые вследствие нарушения процессов рекомбинации в мейозе приводят к утрате или избытку фрагментов хромосом, вовлеченных в перестройку. Частичные моно- или трисомии известны практически по всем хромосомам, но лишь некоторые из них формируют четко диагностируемые клинические синдромы. Фенотипические проявления этих синдромов более полиморфны, чем синдромов целых моно- и трисомии. Отчасти это связано с тем, что размеры фрагментов хромосом и, следовательно, их генный состав, могут варьировать в каждом отдельном случае, а также тем, что при наличии хромосомной транслокации у одного из родителей частичная трисомия по одной хромосоме у ребенка может сочетаться с частичной моносомией по другой. б) Клинико-цитогенетическая характеристика синдромов, связанных с числовыми аномалиями хромосом. 1.Ссиндром Патау (трисомия по хромосоме 13). Впервые описан в 1960 году. Цитогенетические варианты могут быть различны: целая трисомия 13 (нерасхождение хромосом в мейозе, в 80% случаев у матери), транслокационный вариант (робертсоновские транслокации D/13 и G/13), мозаичные формы, дополнительная кольцевая хромосома 13, изохромосомы. Больные имеют тяжелые аномалии строения: расщепление мягкого и твердого неба, незаращение губы, недоразвитие или отсутствие глаз, неправильно сформированные низко посаженные уши, деформированные кости рук и стопы, многочисленные нарушения со стороны внутренних органов, например отмечены врожденные пороки сердца (дефекты перегородок и крупных сосудов). Глубокая идиотия. Продолжительность жизни детей меньше года, чаще 2-3 месяца. Популяционная частота 1 на 7800.
[7] [7]
[5]
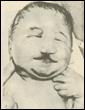
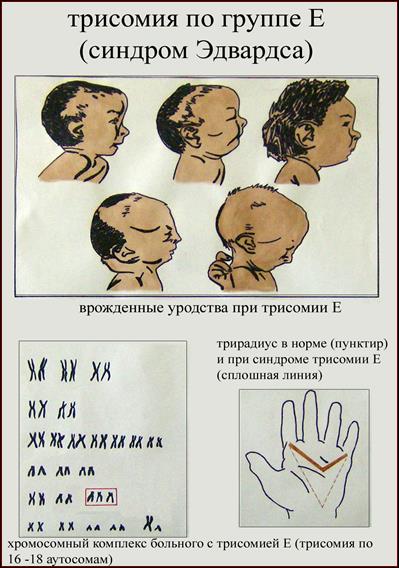
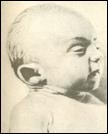
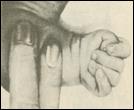
2. Синдром Эдвардса (трисомия по хромосоме 18).. Описан в 1960 году. Цитогенетически в большинстве случаев представлен целой трисомией 18 (гаметическая мутация одного из родителей, чаще по материнской линии). Кроме того, встречаются и мозаичные формы, а транслокации наблюдаются очень редко. Критическим сегментом, ответственным за формирование основных признаков синдрома, является сегмент 18q11. Клинических различий между цитогенетическими формами не обнаружено. Больные имеют узкий лоб и широкий выступающий затылок, очень низко расположенные деформированные уши, недоразвитие нижней челюсти, широкие и короткие пальцы рук. Из
[7]
[5]
внутренних пороков следует отметить комбинированные пороки сердечно-сосудистой системы, незавершенный поворот кишечника пороки развития почек и пр.. Дети с синдромом Эдвардса имеют малую массу тела при рождении. Отмечается задержка психомоторного развития, идиотия и имбецильность. Продолжительность жизни до года - 2-3 месяца. Популяционная частота 1 на 6500. 4. 5. имеют укороченные конечности, маленький череп, плоское и широкое переносье, узкие, с косым разрезом глазные щели, нависающую складку верхнего века – эпикант, избыток кожи на шее, короткие конечности, поперечная четырехпальцевая ладонная складка (обезьянья борозда). Из пороков внутренних органов часто отмечаются врожденные пороки сердца и желудочно-кишечного тракта, которые и определяют продолжительность жизни больных. Характерна психическая отсталость средней степени тяжести. Дети с синдромом Дауна часто ласковые и привязчивые, послушные и внимательные. Жизнеспособность их понижена.
[5]
в) Клинико-цитогенетическая характеристика синдромов, связанных с аномалиями половых хромосом. 1. Синдром Шерешевского-Тернера (моносомия Х-хромосомы). Это единственная форма моносомии у человека, которая может быть
[17]
[5]
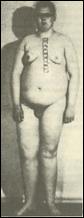
2. Синдром полисомии Х-хромосомы ( Трисомии Х). Цитогенетически выявляются (8) формы 47,ХХХ, 48,ХХХХ и 49,ХХХХХ. С увеличением числа X хромосомы нарастает степень отклонений от нормы. У женщин с тетра- и пентасомией X описаны отклонения в умственном развитии, аномалии скелета и половых органов. Женщины с кариотипом 47,ХХХ в полной или мозаичной форме в основном имеют нормальное физическое и психическое развитие, а интеллект - в пределах нижней границы нормы. У этих женщин имеется ряд нерезких отклонений в физическом развитии, нарушения функции яичников, преждевременный климакс, однако они могут иметь потомство. Популяционная частота 1 на 1000 новорожденных девочек. 3. Синдром Клайнфельтера. Описан в 1942 году. Популяционная частота 1 на 1000 мальчиков. Цитогенетические варианты синдрома могут быть различны: 47.XXY: 48.XXYY; 48.XXXY; 49.XXXXY. Отмечены как полные, так и мозаичные формы. Больные высокого роста с непропорционально длинными конечностями. В детстве отличаются хрупким телосложением, а после 40 лет страдают ожирением. У них развивается астенический или евнуховидный тип телосложения: узкие плечи, широкий таз, жироотложение по женскому типу, слабо развита [5]
мускулатура, скудная растительность на лице. Больные имеют недоразвитие семенников, отсутствие сперматогенеза, снижение полового влечения, импотенция и бесплодие. Обычно развивается умственная отсталость. Коэффициент интеллекта ниже 80. (8) 4. Синдром полисемии Y-хромосомы (дабл-У или «лишней У хромосомы»). Популяционная частота 1 на 1000 мальчиков. Цитогенетически отмечены полные и мозаичные формы. Большинство индивидов по физическому и умственному развитию не отличается от здоровых. Половые железы развиты нормально, рост, как правило, высокий, имеются некоторые аномалии зубов и костной системы. Наблюдаются психопатические черты: неустойчивость эмоций, асоциальное поведение, склонность к агрессии, гомосексуализму. Больные не обнаруживают значительной задержки умственного развития, а часть больных вообще имеет нормальный г) Клинико-цнтогенетнческая характеристика синдромов, связанных со структурными перестройками хромосом. Синдром "кошачьего крика" (моносомия 5р). Описан в 1963 году. Популяционная частота 1 на 50000. Цитогенетические варианты варьируют от частичной до полной делеции короткого плеча хромосомы 5. Для развития основных признаков синдрома большое значение имеет сегмент - 5р15. Кроме простой делеции отмечены кольцевые хромосомы 5, мозаичные формы, а также транслокации между коротким плечом хромосомы 5 (с потерей критического сегмента) и другой аутосомой. Диагностическими признаками заболевания являются: микроцефалия, необычный крик или плач, напоминающий мяуканье кошки (особенно в первые недели после рождения); антимонголоидный разрез глаз, косоглазие, лунообразное лицо, широкая переносица. Ушные раковины низко посажены и деформированы. Имеется поперечная ладонная складка, аномалии строения кистей и пальцев. Умственная отсталость в стадии имбецильносги. Нужно отметить, что такие признаки как лунообразное лицо и кошачий крик с возрастом сглаживаются, а микроцефалия и косоглазие выявляются более отчетливо. Продолжительность жизни зависит от тяжести врожденных пороков развития внутренних органов. Большинство больных погибают в первые годы жизни. д) Клинико-цитогенетическая характеристика синдромов и злокачественных новообразований, связанных с микроструктурными аномалиями хромосом. В последнее время клинико-цитогенетические исследования стали опираться на высокоразрешающие методы хромосомного анализа, позволившее подтвердить предположение о существовании микрохромосомных мутаций, выявление которых находится на грани возможностей светового микроскопа. Используя стандартные цитогенетические методы можно достичь визуального разрешения хромосом с числом сегментов не более 400, а с применением методов прометафазного анализа, предложенного Юнисом в 1976 году, удается получать хромосомы с числом сегментов до 550-850. Незначительные нарушения в структуре хромосом могут быть выявлены с помощью этих методов хромосомного анализа не только среди больных с МВПР, но и при некоторых неизвестных менделирующих синдромах, различных злокачественных образованиях. Большинство синдромов, связанных с микроаномалиями хромосом, встречается редко - 1 случай на 50000-100000 новорожденных. Ретинобластома. Больные с ретинобластомой - злокачественной опухолью сетчатки глаза, составляют 0,6-0,8% от числа всех больных с онкозаболеваниями. Это первая опухоль, для которой установлена связь с хромосомной патологией. Цитогенетически при данном заболевании выявляется микроделеция 13 хромосомы, сегмента 13q14. Кроме микроделеций встречаются мозаичные формы и транслокационные варианты. Описано несколько случаев транслокации сегмента 13 хромосомы на Х-хромосому. Не отмечено корреляции между размерами делецированного фрагмента и фенотипическими проявлениями. Заболевание обычно начинается в возрасте около 1,5 лет и первыми признаками являются свечение зрачков, вялая реакция зрачка на свет, а затем и снижение зрения вплоть до слепоты. Осложнениями ретинобластомы являются отслойка сетчатки, вторичная глаукома. В 1986 году в критическом сегменте 13ql4 обнаружен ген-супрессор опухоли RBI, который явился первым антионкогеном, обнаруженным у человека. е) Моногенные заболевания, проявляющиеся хромосомной нестабильностью. К настоящему времени установлены новые типы изменчивости генома, отличающиеся по частоте и механизмам от обычного мутационного процесса. Одним из проявлений нестабильности генома на клеточном уровне является хромосомная нестабильность. Нестабильность хромосом оценивают по увеличению спонтанной и/или индуцированной частоте хромосомных аберраций и сестринских хроматидных обменов (СХО). Впервые повышенная частота спонтанных хромосомных аберраций была показана в 1964 году у больных с анемией Фанкони, а повышенная частота СХО была обнаружена при синдроме Блюма. В 1.968 году было установлено, что пигментная ксеродерма - фотодерматоз, при котором повышена частота индуцированных УФ-облучением хромосомных аберраций, связана с нарушением способности клеток репарировать (восстанавливать) свою ДНК от повреждений, вызванных УФ-облучением. В настоящее время известно около полутора десятков моногенных патологических признаков, связанных с повышенной ломкостью хромосом. При этих заболеваниях нет специфических участков хромосомных повреждений, однако повышается общая частота аберраций хромосом. Молекулярный механизм данного явления чаще всего связан с дефектами отдельных генов, кодирующих ферменты репарации ДНК. Поэтому большинство болезней, сопровождающихся хромосомной нестабильностью, называют еще болезнями репарации ДНК. Несмотря на то, что по своим клиническим проявлениям эти болезни различны, для всех них характерны повышенная склонность к злокачественным новообразованиям, признаки преждевременного старения, неврологические расстройства, иммунодефицитные состояния, врожденные пороки развития, кожные проявления, нередко наблюдается умственная отсталость. Помимо мутаций генов репарации ДНК в основе болезней с хромосомной нестабильностью могут лежать дефекты и других генов, обеспечивающих стабильность генома. В последнее время накапливается все больше данных о том, что помимо заболеваний, проявляющихся нестабильностью структуры хромосом, имеются и моногенные дефекты, приводящие к болезням с нестабильностью числахромосом. В качестве такой самостоятельной группы моногенных болезней можно выделить редкие патологические состояния, указывающие на неслучайный, наследственно обусловленный характер нерасхождения хромосом в соматических клетках в ходе эмбриогенеза. При цитогенетическом исследовании у этих больных в небольшой части клеток (обычно в 5-20%) выявляется соматический мозаицизм сразу по нескольким хромосомам набора или у одной супружеской пары может быть несколько сибсов с хромосомным мозаицизмом. Предполагается, что такие больные являются "митотическими мутантами" по рецессивным генам, контролирующим отдельные этапы прохождения митоза. Нет сомнения в том, что большинство подобного рода мутаций являются летальными, а выжившие индивиды имеют относительно легкие формы патологии клеточного деления. Несмотря на то, что указанные выше заболевания обусловлены дефектами отдельных генов, проведение цитогенетического исследования у больных с подозрением на данную патологию поможет врачу в дифференциальной диагностике этих состояний. Заболевания с нестабильностью структуры хромосом: Синдром Блюма. Описан в 1954 году. Основными диагностическими признаками являются: низкий вес при рождении, задержка роста, узкое лицо с эритемой в виде бабочки, массивный нос, иммунодефицитные состояния, склонность к злокачественным новообразованиям. Умственная отсталость отмечается не во всех случаях. Цитогенетически характеризуется увеличением числа сестринских хроматидных обменов (СХО) на клетку до 120-150, хотя в норме их число не превышает 6-8 обменов на 1 клетку. Кроме того, с высокой частотой обнаруживаются хроматидные разрывы, а также дицентрики, кольца и хромосомные фрагменты. У больных обнаруживаются мутации в гене ДНК-лигазы 1, локализованном на 19 хромосоме- 19q13.3, однако ген синдрома Блюма картирован в сегменте 15q26.1. Анемия Фанкони. Заболевание с аутосомно-рецессивным типом наследования. Описано в 1927 году. Основные диагностические признаки: гипоплазия лучевой кости и большого пальца, задержка роста и развития, гиперпигментация кожи в паховой и подмышечных областях. Кроме того, отмечаются гипоплазия костного мозга, склонность к лейкозам, гипоплазия наружных половых органов. Цитогенетически характеризуется множественными хромосомными аберрациями - разрывами хромосом и хроматидными обменами. Это генетически гетерогенное заболевание, т.е. клинически сходный фенотип обусловлен мутациями разных генов. Существует по крайней мере 7 форм этого заболевания: A - ген локализован в сегменте 16q24.3; В - локализация гена неизвестна; С - 9q22.3; D - Зр25.3; Е - 6р22; F - 11р15; G (MIM 602956) - 9р13. Наиболее часто встречается форма А - около 60% больных. Синдром Вернера (синдром преждевременного старения). Заболевание с аутосомно-рецессивным типом наследования. Описано в 1904 году. Основными диагностическими признаками являются: преждевременное поседение и облысение, атрофия подкожной жировой клетчатки и мышечной ткани, катаракта, ранний атеросклероз, эндокринная патология (сахарный диабет). Характерны бесплодие, высокий голос, склонность к злокачественным новообразованиям. Больные умирают в возрасте 30-40 лет. Цитогенетически характеризуется клеточными клонами с разными хромосомными транслокациями (мозаицизм по различным транслокациям). Ген заболевания локализован в сегменте 8р11-р12. ж) Синдром ломкой Х-хромосомы. Как правило, разрывы хромосом или пробелы хроматид, возникающие с повышенной частотой в тех или иных конкретных хромосомных сегментах (так называемые ломкие участки или фрагильные сайты хромосом), не связаны с какими-либо заболеваниями. Однако из этого правила есть исключение. В 1969 году у больных с синдромом, сопровождающимся умственной отсталостью, обнаружилось наличие специфического цитогенетического маркера - в дистальной части длинного плеча X-хромосомы в сегменте Xq27.3 в отдельных клетках фиксируется разрыв или пробел хроматид. Позднее было показано, что первое клиническое описание семьи с синдромом, в котором умственная отсталость является ведущим клиническим признаком, был описан еще в 1943 году английскими врачами П. Мартином и Ю. Белл. Синдром Мартина-Белл или синдром, ломкой Х-хромосомы характеризуется ломкой (фрагильной) Х-хромосомой в сегменте Xq27.3, которая выявляется в специальных условиях культивирования клеток в среде с дефицитом фолиевой кислоты. Фрагильный сайт при этом синдроме получил обозначение FRAXA. Основными диагностическими признаками заболевания являются: умственная отсталость, широкое лицо с чертами акромегалии, большие оттопыренные уши, аутизм, гиперподвижность, плохая концентрация внимания, дефекты речи, более выраженные у детей. Отмечаются также аномалии соединительной ткани с гиперрастяжимостью суставов и дефектом митрального клапана. Относительно полный спектр клинических признаков имеют только 60% мужчин с фрагильной Х-хромосомой, 10% больных не имеют лицевых аномалий, 10% имеют только умственную отсталость без других признаков. Синдром фрагильной Х-хромосомы интересен своим необычным наследованием и высокой популяционной частотой (1 на 1500-3000). Необычность наследования состоит в том, что только 80% мужчин, носителей мутантного гена, имеют признаки заболевания, а остальные 20% как клинически, так и цитогенетически нормальны, хотя после передачи мутации своим дочерям могут иметь пораженных внуков. Этих мужчин называют трансмиттерами, т.е. передатчиками неэкспрессированного мутантного гена, который становится экспрессируемым в последующих поколениях. Кроме того, имеется два типа женщин - гетерозиготных носителей мутантного гена: а) дочери мужчин-трансмиттеров, не имеющих симптомов заболевания, у которых не выявляется фрагильная X хромосома; б) внучки нормальных мужчин-трансмиттеров и сестры пораженных мужчин, которые обнаруживают клинические признаки заболевания в 35% случаев. Таким образом, мутация гена при синдроме Мартина-Белл существует в двух формах, отличающихся по своей пенетрантности: первая форма - фенотипически не проявляющаяся премутация, которая переходит в полную мутацию (вторая форма) при прохождении через женский мейоз. Обнаружена четкая зависимость развития умственной отсталости от положения индивида в родословной. При этом хорошо прослеживается явление антиципации - более тяжелого проявления заболевания в последующих поколениях. Молекулярный механизм мутации стал понятен в 1991 году, когда был охарактеризован ген, ответственный за развитие данного заболевания. Ген получил название FMR1 (англ. - Fragile site Mental Retardation 1 - ломкий участок хромосомы, связанный с развитием умственной отсталости 1 типа). Было установлено, что в основе клинических проявлений и цитогенетической нестабильности в локусе Xq27.3 лежит многократное увеличение в первом экзоне гена FMR-1 простого тринуклеотидного повтора CGG. У нормальных людей число этих повторов в Х-хромосоме колеблется от 5 до 52, а у больных их число составляет 200 и более. Такое явление резкого, скачкообразного изменения числа CGG-повторов у больных получило название экспансии числа тринуклеотидных повторов: Показано, что экспансия CGG-повторов существенно зависит от пола потомка, она заметно увеличена при передаче мутации от матери к сыну. Важно отметить, что экспансия нуклеотидных повторов является постзиготическим событием и возникает на очень ранних стадиях эмбриогенеза. Молекулярная цитогенетика Молекулярно-цитогенетические методы, в основе которых лежит использование
|
|||||||||
|
|
Последнее изменение этой страницы: 2016-04-19; просмотров: 824; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.145.166.178 (0.015 с.) |




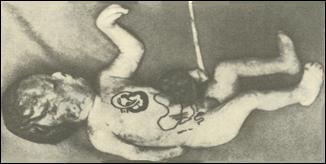
 Синдром Дауна (трисомия хромосомы 21). Впервые описан в 1866 году английским врачом Дауном. Популяционная частота 1 случай на 600-700 новорожденных детей.Частота рождения детей с данным синдромом зависит от возраста матери и резко увеличивается после 35 лет. Цитогенетические варианты очень разнообразны, но около Рис. 15. С. Дауна (6) вверху (8)внизу
Синдром Дауна (трисомия хромосомы 21). Впервые описан в 1866 году английским врачом Дауном. Популяционная частота 1 случай на 600-700 новорожденных детей.Частота рождения детей с данным синдромом зависит от возраста матери и резко увеличивается после 35 лет. Цитогенетические варианты очень разнообразны, но около Рис. 15. С. Дауна (6) вверху (8)внизу 95% случаев представлены простой трисомией 21 хромосомы, в результате нерасхождения хромосом в мейозе у родителей. Наличие полиморфных молекулярно-генетических маркеров позволяет определить конкретного родителя и стадию мейоза в которой произошло нерасхождение. Несмотря на интенсивное изучение синдрома причины нерасхождения хромосом до настоящего времени не ясны. Этиологически важными факторами считаются внутри- и внефолликулярное перезревание яйцеклетки, снижение числа или отсутствие хиазм в первом делении мейоза. Отмечены мозаичные формы синдрома (2%), робертсоновские транслокационные варианты (4%). Около 50% транслокационных форм наследуются от родителей и 50% являются мутациями de novo. Критическим сегментом, ответственным за формирование основных признаков синдрома, является область 21q22. Больные
95% случаев представлены простой трисомией 21 хромосомы, в результате нерасхождения хромосом в мейозе у родителей. Наличие полиморфных молекулярно-генетических маркеров позволяет определить конкретного родителя и стадию мейоза в которой произошло нерасхождение. Несмотря на интенсивное изучение синдрома причины нерасхождения хромосом до настоящего времени не ясны. Этиологически важными факторами считаются внутри- и внефолликулярное перезревание яйцеклетки, снижение числа или отсутствие хиазм в первом делении мейоза. Отмечены мозаичные формы синдрома (2%), робертсоновские транслокационные варианты (4%). Около 50% транслокационных форм наследуются от родителей и 50% являются мутациями de novo. Критическим сегментом, ответственным за формирование основных признаков синдрома, является область 21q22. Больные
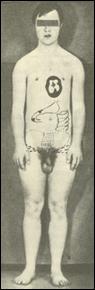


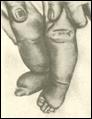

 выявлена у живорожденных. Кроме простой моносомии по X хромосоме, составляющей 50%, встречаются мозаичные формы, делеции длинного и короткого плеча X хромосомы, изо-Х-хромосомы, а также кольцевые X хромосомы. Интересно отметить, что мозаицизм 45,X/46,XY составляет 2-5% от всех больных с этим синдромом и характеризуется широким диапазоном признаков: от типичного синдрома Шерешевского-Тернера до нормального мужского фенотипа. Популяционная частота 1 на 3000 новорожденных. Больные имеют небольшой рост, бочкообразную грудную клетку, широкие плечи, узкий таз, укороченные нижние конечности. Очень характерный признак - короткая шея со складками кожи, идущими от затылка (шея сфинкса). У них наблюдается низкий рост волос на затылке, гиперпигментация кожи, снижение зрения и слуха. Внутренние углы глаз располагаются выше наружных. Часто встречаются врожденные пороки сердца и почек. У больных выявляется недоразвитие яичников. Бесплодны. Интеллектуальное развитие в пределах нормы. Отмечается некоторая инфантильность эмоций, неустойчивость настроения. Больные достаточно жизнеспособны.
выявлена у живорожденных. Кроме простой моносомии по X хромосоме, составляющей 50%, встречаются мозаичные формы, делеции длинного и короткого плеча X хромосомы, изо-Х-хромосомы, а также кольцевые X хромосомы. Интересно отметить, что мозаицизм 45,X/46,XY составляет 2-5% от всех больных с этим синдромом и характеризуется широким диапазоном признаков: от типичного синдрома Шерешевского-Тернера до нормального мужского фенотипа. Популяционная частота 1 на 3000 новорожденных. Больные имеют небольшой рост, бочкообразную грудную клетку, широкие плечи, узкий таз, укороченные нижние конечности. Очень характерный признак - короткая шея со складками кожи, идущими от затылка (шея сфинкса). У них наблюдается низкий рост волос на затылке, гиперпигментация кожи, снижение зрения и слуха. Внутренние углы глаз располагаются выше наружных. Часто встречаются врожденные пороки сердца и почек. У больных выявляется недоразвитие яичников. Бесплодны. Интеллектуальное развитие в пределах нормы. Отмечается некоторая инфантильность эмоций, неустойчивость настроения. Больные достаточно жизнеспособны.
 [7]
[7] интеллект. Могут иметь нормальное потомство в 50% случаев.
интеллект. Могут иметь нормальное потомство в 50% случаев.


