Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Биохимические основы наркоманииСодержание книги
Поиск на нашем сайте
Биохимические основы наркомании Наркотическая зависимость - это хроническое рецидивирующее заболевание, характеризующееся навязчивым поиском и приемом наркотического вещества.
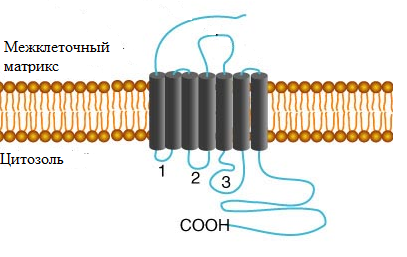
На начальном этапе наркотические вещества вызывают состояние эйфории и подавляют стресс-реакции. Однако, продолжительное их употребление ведет к адаптивным изменениям в центральной нервной системе (ЦНС), формированию физической и психологической зависимости, гипервозбудимости и необходимости постоянного эффекта подкрепления. К группам веществ, обладающих наркотическим действиям относят опиоиды, каннабиноиды, этанол, кокаин, амфетамины, а также никотин. На формирование как алкогольной, так и наркотической зависимости значительное влияние оказывают генетические и социальные факторы. Лица, имеющие биологических родителей, страдающих алкогольной зависимостью, имеют потенциально более сильную склонность к алкоголизму даже в том случае, если они воспитывались в семьях неалкоголиков. Наличие в геноме аллели гена альдегиддегидрогеназы, отвечающей за синтез изофермента с пониженной активностью, снижает вероятность развития алкоголизма, поскольку, в этом случае, употребление алкоголя ведет к накоплению в организме уксусного альдегида, ответственного за формирование симптомов алкогольной интоксикации и, как следствие, снижающего тягу к употреблению алкоголя. Некоторые полиморфизмы гена, ответственного за синтез нейропептида Y и µ-рецептора опиоидов связаны, соответственно, со склонностью к злоупотреблению алкоголем и героином. Подавление активности генов, ответственных за синтез цитохромов семейства Р450 ведет к разобщению метаболизма ксенобиотиков, снижая тем самым риск развития алкогольной и наркотической зависимости, а также тяги к табакокурению. Напротив, полиморфизм гена гидролазы амидов жирных кислот, инактивирующей каннабиноиды, ассоциирован с повышенным влечением к употреблению наркотиков. А1-аллель гена, кодирующего TaqIAD 2-дофаминовый рецептор связана с развитием тяжелого алкоголизма. Рис. 1 Строение ионотропного никотинового ацетилхолинового рецептора (nAChR). Ионотропный рецептор связвает лиганд, что приводит к открытию ионного канала посредством конформационных перестроек в структуре рецептора. рецептор состоит из пяти субъединиц, каждая из которых представлена четырьмя трансмембранными доменами. В отсутствии соответствующего лиганда, данный канал пребывает в закрытом состоянии, не позволяющем ионам проникать через мембрану клетки. Связывание лиганда с рецептором приводит к быстрым конформационным перестройкам в структуре рецептора, приводящим к открытию ионного канала и прохождению через него ионов по электрохимическому градиенту. Данные эффекты реализуются быстро, в течение миллисекунд, и исчезают, как только лиганд диссоциирует из комплекса с рецептором или же, когда происходит десенситизация рецептора по отношению к лиганду. Выделяют два семейства ионотропных рецепторов. Первое семейство включает рецептор ацетилхолина (ACh), никотиновый ацетилхолиновый рецептор (nAChR), рецептор γ-аминомасляной кислоты (ГАМК), рецептор ГАМКА, рецептор глицина, а также один из подклассов серотониновых рецепторов - 5НТ3. Второе семейство объединяет ионотропные рецепторы глутамата. В результате связывания нейромедиатора или наркотика с ионотропным рецептором происходит деполяризация мембраны и активация различных белков. Например, никотин связывается с рецептором nAChR, образующим Na+-канал. Бензодиазепины, барбитураты и этанол связываются с ГАМКА-рецепторами, содержащими Cl--каналы, в результате чего в клетку начинают проникать ионы Cl-. Этанол и фенциклидин ингибируют глутаматные рецепторы, чувствительные к N-метил-D-аспартату (NMDA-рецепторы), сопряженные с Ca2+-и Na+-каналами. 2) Метаботропные рецепторы, в отличие от ионотропных, обычно представлены единственным полипептидом, который передает сигнал внутрь клетки посредством связывания и активации ГТФ-связывающих белков (G-белков). Сигналы, передаваемые посредством метаботропных рецепторов реализуются более длительно (от нескольких десятков секунд до часов), вследствие осуществления большого количества последовательных ферментативных реакций, необходимых для осуществления эффекта. В настоящее время, данную группу рецепторов принято объединять в семейство GPCR (англ. G-protein coupled receptors). Данная группа рецепторов известна также под названием рецепторов семейства Т7, поскольку все рецепторы данного семейства имеют в составе своих молекул 7 трансмембранных доменов. Строение GPCR-рецепторов показано на Рис. 2.
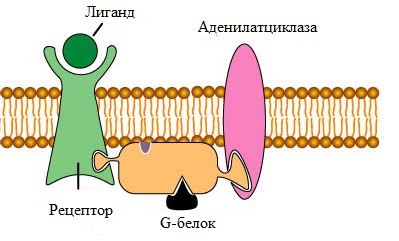
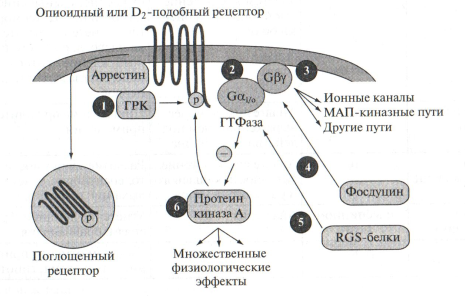
Рис. 2 Строение метаботропного рецептора. Метаботропный рецептор связывает лиганд. При этом в структуре рецептора происходят конформационные перестройки, приводящие к активации G -белка. Последние, в свою очередь, активируют ассоциированные с мембраной ферменты (например аденилатциклазу), генерирующие вторичные посредники в передаче сигнала внутрь клетки (например, цАМФ). К данному классу рецепторов принадлежат опиоидные рецепторы, β-адренергические рецепторы, дофаминовые рецепторы и канабиоидные рецепторы. Метаботропные рецепторы опосредуют медленные реакции. Ассоциированные с ними G-белки представляют собой гетеротримеры, состоящие из α-, β- и γ-субъединиц. α-субъединица обладает ГТФазной активностью. β- и γ-субъединицы, в отсутствие α-субъединицы способны образовывать βγ-димеры, не проявляющие собственной ферментативной активности, однако способные регулировать работу ионных каналов по принципу белок-белковых взаимодействий. В отсутствие лиганда, активный центр α-субъединицы содержит молекулу ГДФ и находится в комплексе с β- и γ-субъединицами. При связывании лиганда с рецептором, происходит активация ассоциированного с ним G-белка, что приводит к диссоциации α-субъединицы от βγ-димера. При этом происходит замещение молекулы ГДФ в активном центре α-субъединицы на молекулу ГТФ: α-ГДФ + ГТФ => α-ГТФ + ГДФ Как α-субъединица, так и βγ-димер могут активировать или ингибировать ферменты (аденилатциклазу или фосфолипазу С), в результате чего активируется (или подавляется) синтез вторичных посредников: цАМФ, инозитол-1,4,5-трифосфата (ИФ3) и диацилглицерола (ДАГ). Кроме того, β- и γ-субъединицы могут напрямую регулировать работу Ca2+, Na+ и K+-ионных каналов. Вторичные посредники также регулируют работу ионных каналов по механизму фосфорилирования/дефосфорилирования, посредством активации протеинкиназ. Активируемые вторичными посредниками протеинкиназы запускают каскадные процессы фосфорилирования различных белков в цитоплазме и ядре клетки, приводя к изменению активности ферментов и уровня экспрессии отдельных генов. Различные наркотические вещества реализуют свои эффекты через многочисленные рецепторы с использованием большого количества сигнальных механизмов. Связывание опиоидов с опиоидными рецепторами приводит к ингибированию аденилатциклазы и, соответственно, снижению уровня цАМФ и активности сАМФ-зависимой протеинкиназы А (ПКА/PKA). Каннабиноиды связываются с каннабиноидными рецепторами. Галлюциногены, такие как ЛСД, кетамин, скополамин и атропин, являются частичными агонистами серотониновых рецепторов. Амфетамины и кокаин активируют различные типы дофаминергических, адренергических и серотонинергических рецепторов и повышают уровень дофамина, норадреналина и серотонина в синапсах (Табл. 1). Рис. 3 Сигнальный механизм действия опиатов в нейронах голубого пятна. С другой стороны, снижение активности ПКА приводит к понижению уровня фосфорилирования ряда других белков, включая факторы транскрипции. Одним из таких факторов является белок CREB (англ. - cAMP-response element binding protein), регулирующий экспрессию многих генов, включая ген, кодирующий аденилатциклазу VIII. Повторный прием опиатов приводит к активации экспрессии обеих изоформ аденилатциклазы и, следовательно, к активации синтеза цАМФ и активности ПКА. Фосфорилирование Na+-зависимых ионных каналов приводит к увеличению проницаемости мембраны нейрона для Na+ и, соответственно, к ее деполяризации и началу процессов возбуждения в нейронах голубого пятна. Данные изменения функциональной активности нейронов лежат в основе формирования устойчивости и зависимости при приеме наркотических препаратов, а также отвечают за развитие абстиненции. В настоящее время, полный спектр сигнальных механизмов, вовлеченных в регуляцию нейрональной активности при воздействии наркотиков остается предметом дискуссий. Известно, что активация аденилатциклазы VIII происходит под влиянием CREB, в то время как механизм активации аденилатциклазы I остается невыясненным. Увеличение образования цАМФ при повторном приеме опиатов, кокаина или же алкоголя происходит также в прилежащем ядре. Прилежащее ядро, являясь составной частью мезолимбической системы, играет важную роль в формировании мотивационных состояний и принимает участие в образовании наркотической зависимости. D1-дофаминовые рецепторы действуют через активацию Gs-белков и активируют синтез цАМФ. Стимуляция этого механизма в N. Acc. определяет функциональную гиперчувствительность D1-дофаминовых рецепторов в результате хронического воздействия наркотиков. В основе формирования такой гиперчувствительности лежит, как полагают, фосфорилирование Na+-ионного канала ПКА. Известно, что хроническое воздействие наркотических веществ вызывает изменения уровня фосфорилирования CREB. Гены опиоидных пептидов, ферментов, принимающих участие в синтезе нейромедиаторов, сигнальных белков и транскрипционных факторов содержат CRE-участки, с которыми связывается регуляторный фактор транскрипции CREB и активирует транскрипцию. Белок CREB состоит из двух субъединиц, содержащих остатки Ser133. При фосфорилировании данных остатков белок CREB приобретает сродство к CRE-участку и присоединяется к нему, повышая тем самым активность связывания РНК-полимеразы с промотором регулируемого гена. Изменение интенсивности фосфорилирования/дефосфорилирования CREB приводит к изменению синтеза опиоидных пептидов и нейромедиаторов, участвующих в реализации воздействия наркотиков на клетки. Увеличение концентрации цАМФ и уровня фосфорилирования CREB приводит у активации синтеза опиоидного пептида динорфина, который экспрессируется в нейронах полосатого тела и прилежащего ядра и связывается с κ-опиоидными рецепторами, локализованными на пресинаптических дофаминсодержащих нервных окончаниях, локализованных в тегментальной области, прилежащей к черной субстанции. В результате происходит подавление высвобождения дофамина, что приводит к развитию угнетенного состояния. Изменение экспрессии CREB в ответ на хронический прием наркотиков регулируется по принципу отрицательной обратной связи. Снижение уровня цАМФ, вызываемое однократным приемом опиатов приводит к подавлению активности ПКА и уровня фосфорилирования CREB. Это приводит к активации экспрессии гена CREB через регуляторный CRE-участок, имеющийся в данном гене. Повышение продукции CREB активирует аденилатциклазу VIII и вызывает накопление цАМФ. Таким образом, в то время как в нейронах увеличивается экспрессия CREB, в них сохраняется низкий уровень цАМФ, поскольку опиаты, при однократном введении ингибируют аденилатциклазу через Gi-сопряженные рецепторы. Активация синтеза цАМФ при повторном введении наркотика представляет собой компенсаторную реакцию, имеющую место, в нейронах, секретирующих динорфин и ГАМК, и иннервирующих дофамин- и серотонинергические нейроны. Увеличение высвобождения ГАМК, вызванное активацией синтеза цАМФ, наблюдается при абстинентном синдроме. Хроническое поступление в организм наркотиков приводит к адаптации клеточных рецепторных механизмов, десенситизации клеток-мишеней и, как следствие к формированию устойчивости к действию препарата и необходимости увеличения его дозы. Одним из механизмов данного эффекта является фосфорилирование рецепторов с участием различных типов протеинкиназ. Фосфорилированный рецептор подвергается интернализации внутрь клетки, нарушается его сопряжение с G-белком, и развивается десенситизация. В процессах фосфорилирования опиоидных и дофаминовых рецепторов принимают участие G-белок-рецепторные протеинкиназы (ГРК) (Рис. 4-1), которые фосфорилируют только те рецепторы, которые связаны с лигандом. Фосфорилирование лиганд-рецепторного комплекса является сигналом для связывания с ним белков "аррестинов", вызывающих изоляцию рецепторов с клеточной поверхности. Еще один возможный механизм связан с насыщением околорецепторной зоны G-белками, их α- (Рис. 4-2) и βγ-субъединицами (Рис. 4-3), а также другими белками, такими как фосдуцин (Рис. 4-4) и RGS-белки (Рис. 4-5), модулирующими функции G-белков. Фосфорилирование рецептора ПКА не может опосредовать десенситизацию рецепторов, потому что активация рецептора ведет к ингибированию ПКА. Однако, увеличение активности ПКА (Рис. 4-6) при хроническом поступлении наркотиков в организм способно инициировать фосфорилирование и, таким образом, регулировать функциональное состояние рецепторов в процессе абстиненции.
Рис. 4 Механизм изменения чувствительности опиоидных и дофаминовых рецепторов к действию наркотиков Другой механизм развития устойчивости может быть связан с вызванными наркотиками модификациями K+- и Ca2+-ионных каналов, регулируемых G-белками. Рис. 5 Воздействие наркотических веществ на экспрессию генов Рис. 8. Размещение субстрата в активном центре алкогольдегидрогеназы При связывании субстрата, его гидроксильная группа сближается с ионом Zn 2+ и утрачивает протон, превращаясь в алкоголят. В отщеплении протона и стабилизации алкоголята участвует система водородных связей с Ser -48 и His -51. Каталитический центр АДГ содержит ион Zn2+, который окружен четырьмя лигандами, расположенными в вершинах неправильного тетраэдра. Три из них принадлежат белку и образованы остатками Cys-46, Cys-174 и His-67. Четвертое координационное положение занимает кислород субстрата, а в его отсутствие - молекула воды (Рис. 8). Гидроксильная группа остатка Ser-48 способна образовывать водородную связь с кислородом лиганда - воды или спирта. Спирт, гидрофобная цепь которого связана в гидрофобном канале фермента, ориентируется так, что его кислород устанавливает непосредственную связь с ионом Zn2+, вытесняя из четвертого координационного положения, находившуюся там молекулу воды (Рис. 8). Это приводит к отщеплению протона гидроксильной группы, а связь с Zn2+ образует алкоголят-анион спирта R-CH2O-:
Алкоголят-анион далее стабилизируется путем смещения электронной плотности от атома кислорода алкоголята к связи С-О, которая в конечном счете превращается в двойную, а гидрид-ион от атома углерода переносится в положение 4 никотинамидного ядра НАД+:
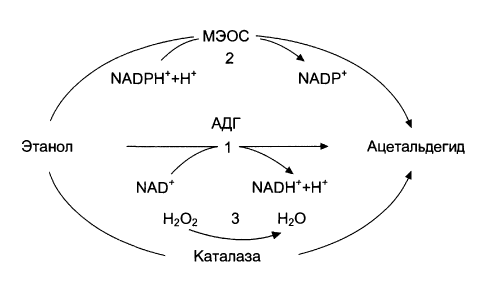
При обратной реакции, перенос гидрид-иона происходит от НАДН к углероду альдегидной группы, двойная связь которой оказывается поляризованной за счет влияния связанного с ферментом иона Zn2+ на атом кислорода. Такая поляризация усиливает частичный положительный заряд на атоме углерода карбонильной группы, что благоприятствует присоединению гидрид-иона. - Микросомальное окисление Данный тип окисления этанола протекает в микросомах клеток печени при участии цитохром Р450-зависимой микросомальной этанолокисляющей системы (МЭОС): С2Н5ОН + НАДФН + Н+ + О2 CH3CHO + НАДФ+ + 2Н2О МЭОС локализована в мембранах гладкого эндоплазматического ретикулума (ЭР) гепатоцитов. Данная система обеспечивает у здоровых людей окисление порядка 10% поступившего в организм этанола. При злоупотреблении алкоголем, а также другими спиртами или же лекарственными препаратами типа барбитуратов, роль данной системы в метаболизме данных веществ существенно возрастает. Этот путь окисления этанола осуществляется с участием изофермента цитохрома Р450 - Р450 IIE1 (Cyp2E1). При хроническом алкоголизме, окисление этанола по данному пути ускоряется на 50-70%, за счет гипертрофии ЭР и индукции Р450 IIE1 (Cyp2E1). Побочными продуктами этой реакции являются активные формы кислорода (АФК), ответственные за окислительное повреждение клеточных структур. Еще одним фактором, способствующим поражению гепатоцитов при хроническом злоупотреблении алкоголем, является употребление и пищей больших количеств полиненасыщенных жирных кислот (ПНЖК). Микросомы играют важную роль в метаболизма ПНЖК. При избытке ПНЖК в диете или при состояниях, характеризующихся высоким уровнем свободных жирных кислот (ЖК) в крови (ожирение, сахарный диабет II типа), повышается ДНК-связывающая активность микросомального пролиферативного α-рецептора (PPAR). Это индуцирует экспрессию генов микросомальных ферментов, Cyp4A1 и Cyp4A2, что приводит к еще более активной генерации АФК и окислительному повреждению клеточных структур. Кроме того, активируется ω-окисление длинноцепочечных ЖК, опосредованное Cyp4A, с образованием токсичных дикарбоновых кислот. Этанол также является субстратом для микросомальной каталазы. - Окисление с помощью каталазы, тканевых оксидаз и пероксидаз Данный механизм играет второстепенную роль в процессе окисления этанола. У здоровых людей он ответственен за окисление 2-3% экзогенного этанола. Окисление протекает согласно следующему уравнению: С2Н5ОН + Н2О2 CH3CHO + 2Н2О У людей, страдающих алкоголизмом роль этого механизма окисления этанола существенно возрастает. Суммарно, пути окисления этанола можно представить следующей схемой:
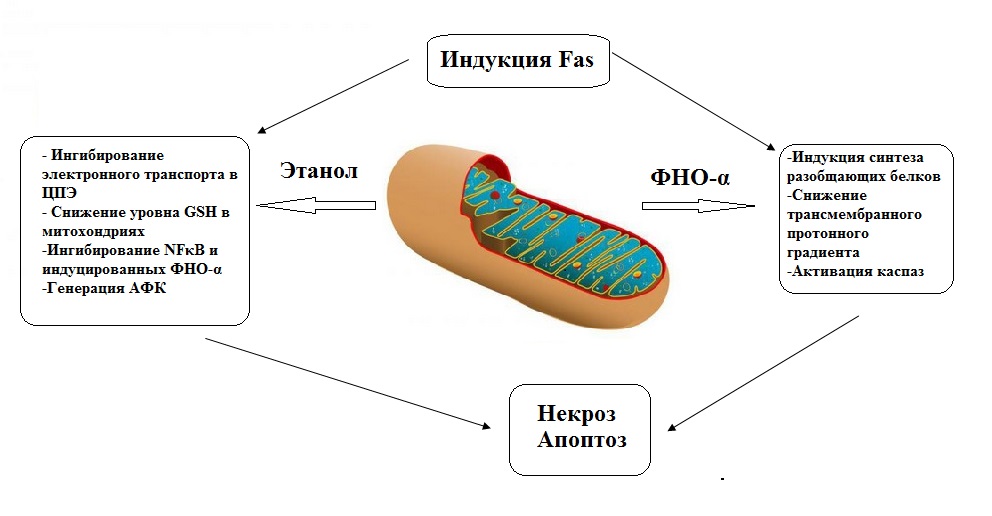
Рис. 9 Метаболические эффекты этанола в печени Реакции 1 - реакция, катализируемая АДГ; 2 - реакция, катализируемая АлДГ; 3 - реакция, катализируемая ацетил-СоА-синтетазой; Скорость образования ацетальдегида (1) при приеме большого количества алкоголя выше, чем скорость его окисления (2), поэтому ацетальдегид накапливается и оказывает влияние на структуру и синтез белков, а также снижает концентрацию восстановленного глутатиона (5), что приводит к активации перекисного окисления липидов (ПОЛ). Скорость глюконеогенеза (6) снижается за счет повышения концентрации НАДН, образующегося при окислении этанола. Лактат (7) выделяется в кровь, что приводит к развитию лактоацидоза. Увеличение концентрации НАДН замедляет скорость ЦТК. В результате происходит накопление ацетил-СоА и активируется синтез кетоновых тел (8). Окисление жирных кислот замедляется (9); увеличивается интенсивность синтеза триглицеридов (10) и холестерина (10), что приводит к ожирению, гипертриацилглицеролемии, и, в конечном счете атеросклерозу, жировому перерождению и циррозу печени. На начальных стадиях алкоголизма, окисление ацетил-СоА в ЦТК является основным источником энергии в клетке. Избыток ацетил-СоА, образовавшегося из этанола, выводится из митохондрий в виде цитрата и используется в цитоплазме для синтеза жирных кислот и холестерина. Этот процесс требует использования АТФ и НАДФН. Последний образуется в ходе двух реакций окислительной фазы пентозофосфатного пути окисления глюкозы. В этих условиях, снижение скорости глюконеогенеза приводит к дефициту эндогенной глюкозы, синтезированной de novo. Возникающий в связи с этим дефицит НАДФН приводит к дисбалансу в системе работы ферментов антиокислительной защиты (глутатионпероксидаза/глутатионредуктаза). НАДФН является коферметном глутатионредуктазы, осуществляющей поддержку пула восстановленного глутатиона: GSH + H2O2 GSSG + 2H2O (ГПО) GSSG + НАДФН+Н+ 2GSH + НАДФ+ С другой стороны, у лиц, страдающих алкогольной зависимостью, нередко наблюдается дефицит тиамина (витамин В1) в организме из-за нарушения режима питания. Кроме того, показано, что алкоголь подавляет всасывание тиамина, поскольку ингибирует кишечную АТФазу, участвующую в абсорбции В1. Поэтому, при введении им глюкозы, может происходить быстрое накопление пирувата и лактата, приводящее к лактоацидозу, нередко со смертельным исходом. С недостаточностью тиамина связывают развитие синдрома Вернике-Корсакова, который обычно включает два состояния: - тяжелое психическое расстройство (расстройство сознания, паралич глазодвигательных мышц, нарушение координации движений (особенно нижних конечностей, энцефалопатия Вернике). - хронический нейропсихопатический синдром, характеризующийся поведенческими расстройствами и нарушениями памяти (корсаковский психоз). Роль витамина В1 в развитии этих нарушений доказана терпевтическим эффектом введения тиамина. Из жирных кислот и глицерол-3-фосфата образуются триацилглицеролы (ТАГ), которые секретируются в крови в составе ЛПОНП. Повышенная секреция ЛПОНП печенью приводит к развитию гипертриацилглицеролемии. При хроническом алкоголизме, снижение интенсивности синтеза фосфолипидов и белков в печени, в том числе, апобелков, входящих в состав ЛПОНП, вызывает накопление ТАГ в гепатоцитах и жировое перерождение печени. В период острой алкогольной интоксикации может возникать недостаток оксалоацетата из-за избыточного образования ацетил-СоА. С другой стороны, происходит замедление реакций ЦТК вследствие увеличения содержания НАДН. В этих условиях, избыток ацетил-СоА используется для синтеза кетоновых тел (ацетоацетат и β-гидроксибутират), что может стать причиной возникновения кетоацидоза. Повышенное образование НАДН и ацетальдегида в ходе окисления этанола приводит к смещению равновесия реакции, катализируемой АДГ в сторону образования этанола. Этанол способен растворяться в липидах клеточных мембран, нарушая при этом их микровязкость и селективные свойства. Это приводит к нарушениям процессов мембранного транспорта, работы ионных каналов, межклеточных контактов и взаимодействия мембранных рецепторов с сигнальными молекулами, что приводит к фатальным сдвигам в ключевых процессах, регулирующих функциональное состояние клеток. Кроме того, этанол способен проходить через клеточные мембраны и проникать в кровь, лимфу и далее в любые ткани организма. Повреждение печени, наблюдаемое при хроническом употреблении алкоголя, может быть не связано напрямую с воздействием этанола или его метаболитов на гепатоциты. Имеются сведения, что данный процесс может происходить опосредованно, и ключевая роль в нем принадлежит факторам, высвобождаемым в ответ на действие этанола и ацетальдегида. В частности, большое значение приписывается бактериальным эндотоксинам кишечного происхождения. Данные эффекты связывают с повышением проницаемости стенки кишечника и нарушением моторики ЖКТ при воздействии избыточного количества этанола, что способствует стазу и избыточному размножению микрофлоры толстого кишечника, а иногда и колонизации нижних отделов тонкого кишечника. Кроме того, кишечные микроорганизмы способны сами окислять этанол с образованием ацетальдегида, повреждающего кишечный эпителий и увеличивающий проницаемость стенки кишечника для бактериальных токсинов. Медиаторами алкогольиндуцированной гепатотоксичности в данном случае выступают цитокины, в частности фактор некроза опухолей (ФНО-α), секреция которого стимулируется бактериальными эндотоксинами. При этом принципиальное значение имеет совместное воздействие этанола и ФНО-α, так как отдельное воздействие каждого из этих факторов не всегда бывает достаточным для гибели гепатоцитов. Эффекты совместного воздействия этанола и ФНО-α могут реализовываться, по меньшей мере, двумя путями. Во-первых, как уже отмечалось выше, хроническое воздействие этанола и его метаболитов вызывает нарушение функций митохондрий, которые являются основной мишенью, как для этанола, так и для ФНО-α. Это приводит к снижению интенсивности окислительного фосфорилирования и генерации избыточных количеств АФК. В основе второго механизма лежит нарушение работы внутриклеточных защитных систем: антиоксидантной и антиапоптотической. При хроническом воздействии алкоголя резко снижается уровень восстановленного глутатиона в клетках. Кроме того, установлено подавляющее действие алкоголя на индукцию белковых факторов NF-κB и bcl-XL, которые предотвращают гибель клетки после воздействия ФНО-α и входят в систему защиты клетки от апоптоза. Таким образом, гибель гепатоцитов происходит от совместного воздействия этанола и ФНО-α либо путем апоптоза, индуцированного ФНО-α, либо путем некроза, которому способствует снижение запасов АТФ в клетках, вызванное разобщением окислительного фосфорилирования. Окислительный стресс оказывает выраженное цитотоксическое влияние через трансмембранный рецептор Fas и его лиганд (FasL). У пациентов с алкогольной болезнью печени отмечается высокий уровень FasL в гепатоцитах, обусловленный генерацией АФК. Поскольку на поверхности гепатоцитов экспрессирован также рецептор Fas, это свидетельствует о том, что клетки печени могут быть вовлечены в апоптоз через пара- или аутокринные механизмы (Рис. 10).
Рис. 10 Механизмы повреждающего действия этанола и ФНО-α на гепатоциты Рис. 12 Механизм воздействия этанола на NMDA и AMPA рецепторы Кроме нейромедиаторов, оказывающих быстрые эффекты на клетки-мишени, значительная роль в регуляции мозговых функций принадлежит малым нейропептидам (нейротропинам), выполняющим функции ростовых факторов. К основным нейротропным факторам относятся фактор роста нервов (NGF: nerve growth factor), нейротрофный мозговой фактор (BDNF: brain-derived neurotrophic factor), нейротрофин-3 (NT-3: neurotrophin-3), NT-4 и глиальный нейротрофный фактор (GDNF: glial-derived neurotrophic factor). Острое воздействие алкоголя подавляет проведение сигналов от нейротропинов, что может объяснять тератогенные эффекты этанола на развитие мозга плода в эмбриональном периоде. При хроническом воздействии алкоголя могут наблюдаться разнонаправленные реакции со стороны нейротропинов. В частности, отмечено увеличение уровня BDNF в мозге при длительном употреблении алкоголя. Влияние этанола на кровь Алкоголь при длительном применении вызывает анемию вследствие образования неполноценных эритроцитов с более короткой продолжительностью жизни, а также происходит торможении эритропоэза. У 82-96% алкоголиков отмечается эритроцитарный макроцитоз, который сохраняется после прекращения приема алкоголя до 2-4 месяцев. При регулярном приеме алкоголя снижается образование тромбоцитов в костном мозге, уменьшается срок их жизни, снижается свертываемость крови и могут происходить длительные кровотечения из слизистых оболочек. При алкоголизме воспалительные заболевания протекают с низким содержанием нейтрофилов в крови. Влияние алкоголя на дыхание Через 10-15 минут после попадания этанола в желудок, он обнаруживается в выдыхаемом воздухе. Полное исчезновение запаха алкоголя из выдыхаемого воздуха наблюдается только через 12-15 часов. Алкоголь, содержащийся в выдыхаемом воздухе вызывает раздражение альвеол, фиброз альвеол, хронический бронхит. Хронический бронхит вследствие повреждения рецепторного аппарата приводит к регуляции работы бронхиол, что провоцирует развитие астмы. Часть I 1. Укажите ферменты, участвующие в метаболизме ацетальдегида: 1. Ацил-СоА синтетаза 2. ФАД-зависимая альдегидоксидаза 3. Каталаза 4. Ацил-СоА дегидрогеназа 5. Альдегиддегидрогеназа 2. Первый этап окисления этанола до ацетальдегида осуществляется ферментами: 1. Микросомальная каталаза 2. Альдегиддегидрогеназа 3. Алкогольдегидрогеназа 4. Супероксиддисмутаза 5. Ацетил-СоА ацилтрансфераза 3. Выберите утверждения, правильно характеризующие свойства альдегиддегидрогеназы: 1. АлДГ характеризуется абсолютной субстратной специфичностью 2. АлДГ характеризуется широкой субстратной специфичностью 3. Митохондриальная изоформа АлДГ обладает более низким сродством к ацетальдегиду, по сравнению с цитозольной изоформой 4. АлДГ характеризуется высоким уровнем полиморфизма 5. В цитозоле преобладает активность АлДГ, в то время как в митохондриях более высока активность АДГ 4. Активность альдегиддегидрогеназы присутствует в: 1. Митохондриях 2. Пероксисомах 3. ЭПС 4. Цитоплазме 5. Ядре 5. Выберите утверждения, правильно характеризующие свойства ацетальдегида: 1. гидрофобное вещество 2. образуется под действием алкогольдегидрогеназы 3. ингибирует НАДН-дегидрогеназу 4. окисляется при участии кофермента НАДФ 5. способен взаимодействовать с SH-группой глутатиона Часть II 1. Выберите утверждения, правильно характеризующие свойства цитохрома Р450: 1. может индуцироваться многими веществами 2. окисляет липофильные молекулы 3. представляет собой простой белок 4. локализован в мембране аппарата Гольджи 5. гемопротеин 2. Субстратами цитохрома Р450 могут быть: 1. экзогенные гидрофильные вещества 2. гидрофобные ксенобиотики 3. экзогенные гидрофобные вещества 4. лекарства 5. эндогенные гидрофильные вещества 3. Изоформы цитохрома Р450 различаются по: 1. первичной структуре 2. субстратной специфичности 3. локализации 4. строению активного центра 5. функции 4. Выберите утверждения, правильно характеризующие свойства ацетальдегида: 1. гидрофобное вещество 2. образуется под действием алкогольдегидрогеназы 3. ингибирует НАДН-дегидрогеназу 4. окисляется при участии кофермента ФАД 5. способен взаимодействовать с SH-группой глутатиона 5. Установите соответствие между пунктами, обозначенными цифрой и буквой:
6. Установите соответствие между названием фермента и ктклизируемой им реакцией:
7. Установите соответствие между названиями наркотических веществ и механизмами их действия:
8. Выберите из списка протеинкиназы, принимающие участие в сигнальных механизмах, вовлеченных в действие на нейроны наркотических веществ: 1. киназа гликогенсинтазы 2. МАПК 3. G-белок-рецепторные протеинкиназы 4. ПКА 5. Фосфофруктокиназа-1 9. Выберите структуры ЦНС, принимающие активное участие в формировании наркотической зависимости: 1. прилежащее ядро 2. голубое пятно 3. паравентрикулярное ядро 4. черная субстанция 5. супраоптическое ядро 10. Выберите типы рецепторов, относящихся к ионотропным: 1. α-адренергический 2. рецептор глутамата 3. ГАМКА-рецептор 4. Ацетилхолиновый рецептор 5. Рецептор дофамина Задача №1 Запишите общую схему метаболизма этанола с участием системы, назовите вещества, участвующие в этой схеме, укажите лимитирующую стадию всего процесса. Задача №2 У пациентов, длительно употребляющих алкоголь, наблюдается снижение эффективности действия лекарств, а также наркотических средств, применяемых при хирургическом вмешательстве. Почему изменяется скорость биотрансформации лекарственных веществ у людей, страдающих алкогольной зависимостью? Для ответа: а) Напишите реакции катаболизма этанола в печени; б) Объясните, как влияет частое употребление алкоголя на активность микросомального окисления в печени, и почему снижается эффективность действия лекарств; в) Укажите компоненты микросомальной системы окисления и ее свойства, лежащие в основе привыкания к алкоголю и лекарствам. Задача №3 Дайте сравнительную характеристику общей системы микросомального окисления (МОС) и микросомальной этанолокисляющей системы (МЭОС). Запишите суммарные уравнения реакций, протекающих с участием этих систем окисления. Задача №4 Врачом скорой помощи был установлен факт смерти мужчины в возрасте 53 лет на дому. Как сообщили родственники, он страдал алкоголизмом, а в последний год у него наблюдалась бессонница. Чтобы заснуть, мужчина принимал снотворные препараты, но обычные дозы ему не помогали. Поэтому, он самовольно, без назначения врача, принимал высокие дозы этих препаратов. Каковы возможные причины смерти пациента? Для ответа: а) Изобразите схему детоксикации этанола и снотворных препаратов (например, барбитуратов) в печени; б) Поясните, из-за чего пациенту приходилось принимать большие дозы снотворных препаратов; в) Предложите, при каком условии прием снотворных препаратов может вызывать летальный исход; Задача №5 Этиленгликоль в печени окисляется с образованием токсичной для организма щавелевой кислоты. Фермент, отвечающий за первую стадию данного процесса называется алкогольдегид
|
||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2021-05-12; просмотров: 417; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.220 (0.212 с.) |

 Использование веществ, обладающих свойством изменять настроение, вызывать чувство эйфории и удовольствия известно с древнейших времен. Одним из наиболее рано вошедших в употребление веществ, обладающих наркотическим действием является этиловый спирт. Производство алкогольных напитков имеет, как минимум 8000 летнюю историю. Патофизиологические механизмы формирования алкогольной и наркотической зависимости имеют немало общих черт и закономерностей. Еще одним издавна употребляемым продуктом наркотического действия является табак, известный в Европе со времен экспедиции Х. Колумба и содержащий растительный алкалоид никотин, способный связываться и активировать ацетилхолиновые рецепторы. В Азиатских странах, на протяжении многих тысячелетий используется конопля.
Использование веществ, обладающих свойством изменять настроение, вызывать чувство эйфории и удовольствия известно с древнейших времен. Одним из наиболее рано вошедших в употребление веществ, обладающих наркотическим действием является этиловый спирт. Производство алкогольных напитков имеет, как минимум 8000 летнюю историю. Патофизиологические механизмы формирования алкогольной и наркотической зависимости имеют немало общих черт и закономерностей. Еще одним издавна употребляемым продуктом наркотического действия является табак, известный в Европе со времен экспедиции Х. Колумба и содержащий растительный алкалоид никотин, способный связываться и активировать ацетилхолиновые рецепторы. В Азиатских странах, на протяжении многих тысячелетий используется конопля.