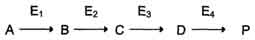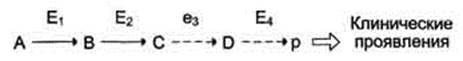Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Воспаление. Нарушение микроциркуляции.
Расстройства кровообращения и микроциркуляции в воспаленной ткани Расстройство кровообращения в воспаленной ткани можно наблюдать под микроскопом на прозрачных тканях экспериментальных животных. Классическими объектами являются препараты языка или брыжейки лягушки, брыжейки крысы и морской свинки. Используют также ткани мочевого пузыря и плавательной перепонки лягушки. Подробное описание расстройств кровообращения в этих тканях при воспалении было сделано Конгеймом и известно в истории изучения воспаления как "опыт Конгейма". Он заключается в следующем: язык или брыжейку лягушки растягивают на пробковом кольце вокруг отверстия на препаровальной доске, которую устанавливают под микроскопом. Фактором, вызывающим воспаление, является часто уже само приготовление препарата. Повреждение ткани можно вызвать также, положив на нее кристаллик поваренной соли. Под малым увеличением легко наблюдать процесс расширения артериол, капилляров и венул, маятникообразные движения крови и стаз. Под большим увеличением отмечаются процессы прилипания лейкоцитов к стенке кровеносных сосудов и эмиграции их в воспаленную ткань (рис. 14). В настоящее время для изучения расстройств микроциркуляции при воспалении у теплокровных животных вживляют прозрачные пластинки в серозные полости, используют методы микроскопии терминальных сосудов защечного мешка хомячка, мигательной перепонки глаза кролика и пр. Широко используются микросъемки, инъекции сосудов коллоидными и флюоресцирующими красками. Широко применяются методы введения меченных изотопами белков и других веществ. Расстройства кровообращения в воспаленной ткани развиваются в виде следующих четырех стадий: кратковременное сужение артериол (наблюдается не всегда); расширение капилляров, артериол и венул - элементы активной или артериальной гиперемии; застой крово- и лимфообращения в воспаленной ткани - элементы пассивной, или венозной, гиперемии; остановка кровообращения в воспаленной ткани - стаз. Перечисленные стадии и наблюдаемые при них элементы различных нарушений кровообращения и микроциркуляции в воспаленной ткани не всегда проявляются в типичной форме и в указанной последовательности. Например, при остром воспалении от легкого ожога расстройство кровообращения ограничивается признаками артериальной гиперемии. Сильный ожог кислотой может сразу привести к картине полного стаза. При хроническом воспалении, например при некоторых видах экземы, в ткани часто наблюдаются явления застойной гиперемии и отека, воспаленная ткань синюшна.
В настоящее время есть основания считать, что расстройства микроциркуляции при воспалении качественно отличаются от таковых при артериальной или венозной гиперемиях невоспалительного происхождения. Эти отличия позволяют выделить воспалительную гиперемию как специальный вид нарушений микроциркуляции (А. Д. Адо, Г. И. Мчедлишвили). Особенности воспалительной гиперемии по сравнению с другими формами полнокровия представлены в табл. 15 [показать]. Кратковременное сужение артериол при воспалении вызывается раздражением сосудосуживающих нервов и гладкомышечных клеток артериол повреждающими агентами, которые вызывают воспаление. Сужение артериол является кратковременным потому, что первичное раздражающее действие быстро проходит. Медиатор симпатической иннервации артериол - норадреналин - разрушается моноаминоксидазой, количество которой увеличивается в воспаленной ткани. Далее развивается расширение артериол, капилляров и венул, сопровождающееся ускорением тока крови - артериальная гиперемия. Стадия артериальной гиперемии характеризуется: расширением артериол, капилляров и венул [показать]; ускорением тока крови в сосудах воспаленной ткани [показать]; повышением кровяного давления в капиллярах и венулах. Вследствие расширения артериол и усиления притока крови давление крови в капиллярах и венах воспаленной ткани увеличивается. Застой крови возникает по мере нарастания воспалительного процесса, когда затрудняется отток крови в венозную систему. Существует несколько факторов, способствующих появлению признаков застоя крови в ходе развития воспаления. Факторы эти следующие: Внутрисосудистые факторы [показать]; Внесосудистые факторы [показать]; Стаз - местная остановка кровотока в микроциркуляторном русле, чаще всего в капиллярах. Изменения кровотока во время развития стаза заключаются в следующем [показать].
Перед остановкой кровообращения в сосудах воспаленной ткани могут возникать своеобразные, синхронные с ритмом сердечных сокращений изменения направления токов крови. Они называются маятникообразными движениями крови: в момент систолы кровь движется в капиллярах воспаленной ткани в обычном направлении - от артерий к венам, а в момент диастолы направление крови становится обратным - от вен к артериям. Механизм маятникообразных движений крови в воспаленной ткани состоит в том, что во время систолы пульсовая волна проскакивает через расширенные артериолы и создает картину, известную под названием капиллярного пульса. В момент диастолы кровь встречает препятствия к оттоку по венозной системе и отливает обратно вследствие падения кровяного давления в капиллярах и артериолах во время диастолы. От маятникообразных движений крови в воспаленной ткани следует отличать передвижения крови из одной сосудистой территории в другую под влиянием прорыва тромбов, открытия или закрытия просвета капилляров вследствие их сдавления, регионарного расширения, закупорки агломерированными форменными элементами и других факторов перераспределения крови внутри сосудисто-капиллярной сети воспаленной ткани. Эти перемещения масс крови из одной сосудистой территории в другую в очаге воспаления чаще возникают в стадии застоя крови и наблюдаются в виде потоков крови по капиллярам, не синхронных с сердечными сокращениями, как при маятникообразных движениях. Повреждение капилляров и венул в начале воспалительного процесса вызывает раннюю реакцию тромбоцитов крови, которые прилипают и скапливаются в местах повреждения. Этот процесс, с одной стороны, является защитным, так как "заклеивает" дефектную структуру эндотелиальной стенки, с другой стороны, он является вредным, так как организует в дальнейшем развитие прилипания и выхождение лейкоцитов в воспаленную ткань, т. е. организует воспаление как вредную для организма патологическую реакцию. Этот диалектически противоположный процесс "защитного" и патологического продолжается далее во всех стадиях развития воспаления. В настоящее время получены данные о том, что при повреждении эндотелия капилляров и вен освобождается вещество (медиатор), которое увеличивает "клейкость" внутренней поверхности эндотелия по отношению к тромбоцитам и лейкоцитам. Этот процесс способствует возникновению "краевого стояния" лейкоцитов при воспалении. Природа этого медиатора пока не определена. Возможно, что оно относится к кининам (пептидам). 43. Исходы воспаления. Механизмы образования грануляционной ткани. Исходы воспаления: 1) возврат к нормальному состоянию с сохранением анатомических и функциональных свойств ткани вследствие восстановления специфических элементов (restitutio ad integrum); 2) образование рубцовой ткани, которая может не отразиться на функциональных свойствах органа (например, при небольших рубцах на коже и в других местах) или же вызвать при значительном развитии смещение органов (например, в полости груди при плевритах и перикардитах) и функциональные нарушения, например при образовании рубцов в центральной нервной системе; 3) гибель ткани, а вслед за этим, в зависимости от характера воспаления, места его развития в жизненно важных органах, иногда и гибель всего организма.
Восстановительные процессы в воспаленной ткани Роль соединительнотканных клеток. В зависимости от вида воспаления ткань всегда в большей или меньшей степени разрушается. Это разрушение достигает наибольших размеров при гнойном воспалении. После того как гнойник прорывается или вскрывается хирургическим путем, из него вытекает или удаляется гной, а на месте бывшего воспаления остается полость. В дальнейшем эта полость, или дефект ткани, вызванный воспалением, постепенно восполняется за счет размножения местных соединительнотканных клеток - гистиоцитов и фибробластов. Гистиоциты (макрофаги по И. И. Мечникову), а также моноциты крови дольше сохраняются в очаге воспаления, чем нейтрофилы и другие гранулоциты. Более того, продукты распада в воспаленной ткани, вызывающие гибель гранулоцитов, оказывают стимулирующее влияние на фагоцитарную активность макрофагов. Макрофаги поглощают и переваривают продукты распада в воспаленной ткани, оставшиеся после истечения или удаления гноя. Они очищают воспаленную ткань от этих продуктов распада путем внутриклеточного переваривания. Одновременно среда воспаленной ткани оказывает стимулирующее влияние на размножение этих клеток и метаплазию их в фибробласты и фиброциты. Они образуют таким путем новую, молодую, богатую кровеносными сосудами грануляционную ткань, которая постепенно превращается в волокнистую ткань, называемую рубцом (рис. 18). Важно отметить, что разрушение, вызванное воспалением в различных органах и тканях, например в мозгу, миокарде, никогда не приводит к восстановлению дифференцированных паренхиматозных клеток воспаленного органа. На месте бывшего ранее гнойника образуется соединительнотканный рубец. Это часто приводит ко многим вторичным осложнениям, связанным с постепенным рубцовым стягиванием, к "спайкам", деформирующим нормальную структуру органа и нарушающим его функцию. Хорошо известно вредоносное влияние рубцового спаечного процесса после воспаления в брюшине, после ранения нервных стволов, ранения или воспаления сухожилий, суставов и многих других органов. Грануляционная ткань образуется за счет размножения гистиоцитов и фибробластов, со стороны здоровой ткани в нее врастают кровеносные сосуды. Грануляционная ткань весьма богата водой, ее коллоиды находятся в состоянии гидратации. Грануляционная ткань - это защитный барьер против инфекции. При попытке инфицирования кролика палочкой сибирской язвы через грануляционную ткань все микробы поглощались клетками соединительной ткани, и заражение не наступало. При заражении той же дозой палочки сибирской язвы под кожу здоровый кролик погибал через несколько часов.
После заполнения грануляционной тканью раны в ней начинают происходить изменения. Кровеносные сосуды затягиваются, клетки постепенно разрушаются и рассасываются. Остаются только волокна субстанции соединительной ткани, образующие рубец. Эпителизация раны после заживления вторичным натяжением не происходит, и рубец (например, на коже) остается видимым многие годы. 44.. Мутации, причины, виды мутаций Причины: Мутагены. Классификация мутагенов. Мутагены (равно и вызываемые ими мутации) классифицируют по происхождению (источнику) на эндогенные и экзогенные, а по природе на физические, химические и биологические. Экзогенные мутагены. Их большинство, к ним относятся различные и многочисленные факторы внешней среды (например, радиационное излучение, алкилирующие агенты, окислители, многие вирусы). Эндогенные мутагены образуются в процессе жизнедеятельности организма (например, мутации могут возникать под влиянием свободных радикалов, продуктов липопероксидации). Физические мутагены — ионизирующее излучение и температурный фактор: - ионизирующее излучение (например, а-, (3-, у-лучи, рентгеновское излучение, нейтроны); - радиоактивные элементы (например, радий, радон, изотопы калия, углерода и т.д. — источники ионизирующего излучения); - УФ-излучение; - чрезмерно высокая или низкая температура. Химические мутагены — самая многочисленная группа мутагенов. К химическим мутагенам относятся: - сильные окислители или восстановители (например, нитраты, нитриты, активные формы кислорода); - алкилирующие агенты (например, йодацетамид); - пестициды (например, гербициды, фунгициды); - некоторые пищевые добавки (например, ароматические углеводороды, цикламаты); - продукты переработки нефти; - органические растворители; - Л С (например, цитостатики, содержащие ртуть средства, иммунодеп-рессанты); - другие химические соединения. Биологические мутагены: - вирусы (например, кори, краснухи, гриппа); - Аг некоторых микроорганизмов. Виды мутаций. По причине, вызвавшей мутации их дифференцируют на «спонтанные» и индуцированные.
«Спонтанные» мутациивозникают под влиянием естественных мутагенов экзо- или эндогенного происхождения, без специального (целенаправленного) вмешательства человека. Такие мутации возникают, например, в результате действия химических веществ, образующихся в процессе метаболизма; воздействия естественного фона радиации или УФ-излучения; ошибок репликации и т.д. Индуцированные мутациивызываются направленным воздействием факторов внешней или внутренней среды. Индуцированный мутационный процесс, в свою очередь, может быть контролируемым или неконтролируемым.Контролируемые мутациивызывают целенаправленно, например, в эксперименте с целью изучения механизмов мутагенеза и/или его последствий.Неконтролируемые мутацииразвиваются случайно, например при выбросе радиоактивных элементов в среду обитания при авариях на атомных электростанциях, военных объектах или в экспериментальных лабораториях. По виду клетки, в воторой произошла мутация выделяют гаметические и соматические мутации. Гаметические мутациивыявляются в половых клетках. Они наследуются потомками и, как правило, обнаруживаются во всех клетках организма. Соматические мутациипроисходят в неполовых — соматических клетках организма и проявляются только у того индивида, у которого они возникают. Эти мутации передаются только дочерним соматическим клеткам при их делении и не наследуются следующим поколением индивида. Если соматическая мутация возникает на ранних стадиях дробления зиготы (но не первого ее деления), возникает несколько клеточных линий с различными генотипами (клеточная «мозаика»). Чем раньше в онтогенезе происходит соматическая мутация, тем больше клеток содержит такую мутацию. Подобные организмы получили название мозаичных. У человека мозаицизм наиболее характерен для половых хромосом. По биологическому значению выделяют патогенные, нейтральные и благоприятные виды мутаций. Патогенные мутацииприводят либо к гибели эмбриона (или плода), либо к развитию наследственных и врожденных заболеваний. Нейтральные мутацииобычно не влияют на жизнедеятельность организма (например, мутации, вызывающие веснушки, изменение цвета волос, радужной оболочки глаза). Благоприятные мутацииповышают жизнеспособность организма или вида (например, темная окраска кожного покрова у жителей африканского континента). По масштабу изменений генетического материала мутации выделяют генные и хромосомные или геномные мутации. Генные (точковые) мутациипредставляют собой изменения молекулярной структуры ДНК. Некоторые из этих изменений не оказывают влияния на функцию соответствующего полипептида (например, замена нуклеотидов, не приводящая к замене аминокислоты в силу вырожденности генетического кода). Значительная часть точковых мутаций нарушает «функционирование» гена и приводит к развитию генных (моногенных) болезней. Фенотипически генные болезни наиболее часто проявляются признаками нарушений метаболизма (например, фенилкетонурия, нейрофиброматоз, муковисцидоз, мышечная дистрофия Дюшенна–Беккера). Хромосомные мутации(аберрации) характеризуются изменением структуры отдельных хромосом, агеномные — их числа. 45. Хромосомные аберрации.. Различают внутрихромосомные, межхромосомные и изохромосомные аберрации. Внутрихромосомные аберрацииобнаруживаются в пределах только одной хромосомы. К ним относят делеции, инверсии и дупликации. Делеция представляет собой утрату одного из участков хромосомы (внутреннего или терминального), что может стать причиной нарушения эмбриогенеза и формирования множественных аномалий развития (например, делеция в регионе короткого плеча хромосомы 5, обозначаемая как 5р-, приводит к недоразвитию гортани, ВПР сердца, отставанию умственного развития). Этот симптомокомплекс обозначен как синдром кошачьего крика, поскольку у больных детей из-за аномалии гортани плач напоминает кошачье мяуканье. Инверсиязаключается во встраивании фрагмента хромосомы на прежнее место после его поворота на 180 °. В результате нарушается порядок расположения генов. Дупликацияозначает удвоение (или умножение) какого-либо участка хромосомы (например, трисомия по короткому плечу хромосомы 9 приводит к появлению множественных ВПР, включая микроцефалию, задержку физического, психического и интеллектуального развития). Межхромосомные аберрациихарактеризуются обменом фрагментами между негомологичными хромосомами. Эти аберрации получили названиетранслокаций.Различают 3 варианта транслокаций:реципрокные(обмен фрагментами двух хромосом),нереципрокные (перенос фрагмента одной хромосомы на другую),робертсоновские (соединение двух акроцентрических хромосом в районе их центромер с потерей коротких плеч; в результате образуется одна метацентрическая хромосома вместо двух акроцентрических). Изохромосомные аберрациихарактеризуются образованием одинаковых, но зеркально расположенных фрагментов двух разных хромосом, содержащих одни и те же наборы генов. Это происходит в результате поперечного разрыва хроматид через центромеры (отсюда другое их название — центрическое соединение). 46. Хромосомные болезни, связанные с нарушением числа аутосом и половых хромосом. Инициальное звено патогенеза — геномная или хромосомная мутация. Хромосомный дисбаланс приводит к остановке либо нарушению эмбрионального развития, в т.ч. ранних этапов органогенеза. В результате формируются множественные ВПР. Тяжесть нарушений обычно коррелирует со степенью хромосомного дисбаланса: чем больше хромосомного материала вовлечено в аберрацию, тем раньше проявляется хромосомный дисбаланс в онтогенезе, тем значительнее нарушения физического и психического развития индивида. Как правило, потеря хромосомы или ее части приводит к более тяжелым клиническим последствиям, чем присоединение хромосомы или ее части. Хромосомные болезни классифицируют (рис. 4-14) по критериям изменения структуры и числа хромосом, а также в зависимости от типа клеток (половые или соматические). Ы верстка! вставить рисунок «рис-4-14» Ы Рис. 4-14. Виды хромосомных болезней. Большинство геномных мутаций (полиплоидии, трисомии по крупным хромосомам, моносомии по аутосомам) летальны. Насчитывают сотни болезней, вызванных нарушением структуры хромосом в результате делеции, дупликации, инверсии или транслокации их отдельных участков. Их клиническая картина и тяжесть определяются характером перестройки, величиной вовлеченных фрагментов и их функциональной значимостью. Мутации в гаметах приводят к развитию т.н. полных форм хромосомных болезней, когда изменения кариотипа выявляют во всех клетках организма. Мутации в соматических клетках на ранних этапах эмбриогенеза приводят к развитию мозаицизма — часть клеток организма имеет нормальный кариотип, а другая часть — аномальный. Это вызывает т.н. мозаичные формы хромосомных болезней. Варианты мозаичных организмов могут быть самыми разнообразными: не только из двух, но из трех и более клонов клеток с разными их количественными соотношениями. Фенотипические отклонения от нормы зависят от доли клеток различных типов, т.е. от стадии развития, на которой произошла мутация. Для хромосомных болезней характерно нарушение репродуктивной функции. Трисомии Синдром Патау, Трисомия 13 выявляется с частотой 1:6000. Летальность высокая: более 96% больных погибают в возрасте до 1–1,5 лет. Проявляется заболевание снижением массы тела, микроцефалией, недоразвитием мозга, аномалиями лица (запавшая переносица, расщелина верхней губы и неба), полидактилией, ВПР внутренних органов (поджелудочной железы, селезенки, сердца). Синдром Эдвардса, Трисомия 18 выявляется у 1 из 7000 новорожденных. Около 2/3 детей с синдромом Эдвардса умирают в первые 6 мес жизни. Проявляется синдром Эдвардса сниженной массой тела, аномалиями лицевого и мозгового черепа (долихоцефалией, деформациями ушных раковин, гипоплазией нижней челюсти, микростомией), короткой грудиной, узкими межреберными промежутками, короткой и широкой грудной клеткой, ВПР сердца и других внутренних органов, нарушениями психомоторного развития. Синдром Дауна, Трисомия 21 встречается с частотой 1:750 новорожденных и цитогенетически характеризуется простой трисомией (96% всех случаев болезни), транслокацией акроцентрических хромосом (3%), мозаицизмом (1%). Характерна малая средняя продолжительностью жизни (35 лет). Проявляется заболевание аномалиями лицевого и мозгового черепа (уплощенный затылок; запавшая спинка носа; косой «монголоидный» разрез глаз; толстые губы; утолщенный язык с глубокими бороздами; маленькие, низко расположенные уши; высокое небо), гипотонией мышц, аномалиями развития внутренних органов (сердца, почек, кишечника), короткими пальцами; аномалиями дерматоглифики (поперечная ладонная складка), умственной отсталостью разной степени (от минимальной дебильности до тяжелой идиотии). Моносомии 1:100 000 родившихся детей). Пример: синдром кошачьего крика (5р-), развивающийся в результате делеции части короткого плеча хромосомы 5. Проявления: плач новорожденного, похожий на мяуканье кошки (причиной являются аномалии гортани в виде ее сужения, отечности слизистой оболочки и уменьшения величины надгортанника), черепно-лицевые аномалии (микроцефалия, антимонголоидный разрез глаз, гипертелоризм, круглое лицо у новорожденных, узкое, вытянутое — у взрослых), отставание умственного и физического развития у детей, идиотия у взрослых (у 1% IQ менее 20), нарушения структуры ребер и позвонков.-Частичные моносомии характеризуются делецией части какой-либо хромосомы и встречаются редко (примерно 1:50 000 Аномалии половых хромосом Нарушение расхождения половых хромосом приводит к образованию аномальных гамет: у женщин — XX и 0 (в последнем случае гамета не содержит половых хромосом); у мужчин — XY и 0. При слиянии половых клеток в подобных случаях возникают количественные нарушения половых хромосом. При болезнях, вызванных дефицитом или избытком Х хромосом, нередко наблюдается мозаицизм (таблица 4-1). Синдром Клайнфелтера Частота синдрома: 2–2,5 на 1000 новорожденных мальчиков. В кариотипе могут быть разнообразные цитогенетические варианты (47,XXY; 48,XXXY; 49,XXXXY и др.). Чаще встречается классический вариант 47,XXY. Проявляется патология высоким ростом, непропорционально длинными конечностями, отложением жира по женскому типу, евнухоидным телосложением, скудным оволосением, гинекомастией, гипогенитализмом, бесплодием (в результате нарушения сперматогенеза, снижения продукции тестостерона и увеличения продукции женских половых гормонов), снижением интеллекта (чем больше в кариотипе добавочных хромосом, тем более выражено). Лечение патологии мужскими половыми гормонами направлено на коррекцию вторичных половых признаков. Однако и после терапии больные остаются бесплодными. Синдромы полисомии Х Трисомия Х. Наиболее частым синдромом из группы полисомий X является синдром трисомии Х (47,XXX): частота — 1:1000 новорожденных девочек, кариотип 47,XXX; пол — женский, фенотип женский; как правило, физическое и психическое развитие у женщин с этим синдромом не имеет отклонений от нормы. Синдром Шерешевского–Тернера Частота синдрома равна в среднем 1:3000 новорожденных девочек; кариотип: 45, Х0. Встречаются и другие варианты (например, изохромосома длинного плеча X — Xqi, делеция короткого плеча — Xp, делеция длинного плеча — Xq). Проявляется заболевание низким ростом, короткой шеей с избытком кожи или крыловидной складкой, широкой, часто деформированной грудной клеткой, деформацией локтевых суставов, недоразвитием первичных и вторичных половых признаков, бесплодием. У новорожденных почти во всех случаях наблюдается лимфатический отек кистей и стоп. Раннее лечение женскими половыми гормонами может оказаться эффективным. 47. Наследственные болезни, сцепленные с полом. К важным особенностям доминантного типа наследования заболеваний, сцепленных с полом относят: поражение лиц мужского и женского пола, но женщин в 2 раза чаще; при этом у мужчин отмечается более тяжелое течение заболевания;Ú передача болеющим мужчиной патологического аллеляÚ всем дочерям итолько дочерям,но не сыновьям, поскольку сыновья получают от отца хромосому Y; передача больной женщиной заболевания и сыновьям и дочерям с равной вероятностью.Ú Примерами заболеваний с доминантным X–сцепленным типом наследования могут служить одна из форм гипофосфатемии с доминантным X-сцепленным типом наследования, витамин D-резистентный рахит, болезнь Шарко–Мари–Тута X-сцепленная доминантная, рото-лице-пальцевой синдром I типа. Родословная с таким типом наследования витамин D-резистентного рахита в 4 поколениях представлена на рисунке 4-10. Сцепленное с хромосомой Xрецессивное наследование К числу наиболее значимых признаков заболевании с рецессивным наследованием, сцепленным с Х хромосомой относят следующие: больные дети рождаются в браке фенотипически здоровых родителей;Ú заболевание наблюдается почти исключительно у лиц мужского пола, а матери больных являются облигатными носительницами патологического гена;Ú сын никогда не наследует заболевание от отца;Ú у носительницы мутантного гена вероятность рождения больного ребенка равна 25% (независимо от пола новорожденного), вероятность рождения больного мальчика — 50%.Ú Примерами заболеваний с рецессивнымX-сцепленным типом наследования могут быть: гемофилия A, гемофилия B, X-сцепленная рецессивная болезнь Шарко–Мари–Тута, дальтонизм, мышечная дистрофия Дюшенна–Беккера, синдром Калльмана, болезнь Хантера (мукополисахаридоз типа II), гипогаммаглобулинемия Брутона. Родословная с этим типом наследования (гемофилии А) в 4 поколениях представлена на рисунке 4-11. Голандрический, или сцепленный с хромосомой Y тип наследования Особенностями наследования патологиисY-сцепленным типом считают: передача признака от отца всем сыновьям и только сыновьям;Ú дочери никогда не наследуют признак от отца, т.к. у них нет хромосомы Y;Ú «вертикальный» характер наследования признака;Ú 100% вероятность наследования для лиц мужского пола равна;Ú гены, ответственные за развитие патологического признака, локализованы в хромосоме Y.Ú Примеры признаков, передающихся по Y-сцепленному типу наследования: гипертрихоз ушных раковин, избыточный рост волос на средних фалангах пальцев кистей, азооспермия. Родословная сY-сцепленным типом наследования избыточного оволосения ушных раковин в четырех поколениях представлена на рисунке 4-12. Энзимопатии, /фенилкетонурия/ и их патогенез. Фенилкетонурия Все формы фенилкетонурии — результат недостаточности ряда ферментов. Их гены транскрибируются в гепатоцитах и наследуются по аутосомно-рецессивному типу. Наиболее частая форма фенилкетонурии возникает при мутациях гена фенилаланин 4-монооксигеназы (фенилаланин 4-гидроксилаза, фенилаланиназа). Самый распространенный тип мутаций — однонуклеотидные замены (миссенс-, нонсенс-мутации и мутации в сайтах сплайсинга). Ведущее патогенетическое звенофенилкетонурии — гиперфенилаланинемия с накоплением в тканях токсических продуктов метаболизма (фенилпировиноградной, фенилуксусной, фенилмолочной и других кетокислот). Это ведет к поражению ЦНС, нарушению функции печени, обмена белков, липо- и гликопротеинов, метаболизма гормонов. Проявляется фенилкетонурияповышенной возбудимостью и гипертонусом мышц, гиперрефлексией и судорогами, признаками аллергического дерматита, гипопигментацией кожи, волос, радужки; «мышиным» запахом мочи и пота, задержкой психомоторного развития. У нелеченых детей формируется микроцефалия и умственная отсталость. С этим связано другое название заболевания — фенилпируватная олигофрения. Лечение фенилкетонурии проводят с помощью диетотерапии (исключением или снижением содержания в пище фенилаланина). Диету необходимо соблюдать с момента установления диагноза (первые сутки после рождения) и контролировать содержание фенилаланина в крови не менее 8–10 лет. ЭНЗИМОПАТИИ В основе многих заболеваний лежат нарушения функционирования ферментов в клетке - энзимопатии. Различают первичные (наследственные) и вторичные (приобретённые) энзимопатии. Приобретённые энзимопатии, как и вообще протеинопатии, по-видимому, наблюдают при всех болезнях. При первичных энзимопатиях дефектные ферменты наследуются, в основном, по аутосомнорецессивному типу. Гетерозиготы, чаще всего, не имеют фенотипических отклонений. Первичные энзимопатии обычно относят к метаболическим болезням, так как происходит нарушение определённых метаболических путей. При этом развитие заболевания может протекать
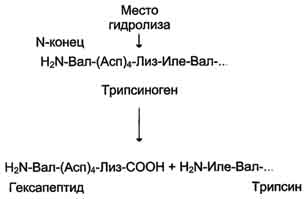
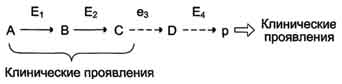
Рис. 2-34. Активация трипсина частичным протеолизом. Под действием фермента кишечника энтеропептидазы происходит гидролиз пептидной связи Лиз-Иле. В результате отщепления гексапептида с N-конца формируется активный центр в оставшейся части молекулы. по одному из ниже перечисленных "сценариев". Рассмотрим условную схему метаболического пути:
Вещество А в результате последовательных ферментативных реакций превращается в продукт Р. При наследственной недостаточности какого-либо фермента, например фермента Е3, возможны разные нарушения метаболических путей: Нарушение образования конечных продуктов. Недостаток конечного продукта этого метаболического пути (Р) (при отсутствии альтернативных путей синтеза) может приводить к развитию клинических симптомов, характерных для данного заболевания:
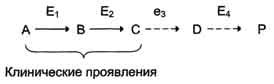
Клинические проявления. В качестве примера можно рассмотреть альбинизм. При альбинизме нарушен синтез в меланоцитах пигментов - меланинов. Меланин находится в коже, волосах, радужке, пигментном эпителии сетчатки глаза и влияет на их окраску. При альбинизме наблюдают слабую пигментацию кожи, светлые волосы, красноватый цвет радужки глаза из-за просвечивающих капилляров. Проявление альбинизма связано с недостаточностью фермента тирозингидроксилазы (тирозиназы) - одного из ферментов, катализирующего метаболический путь образования меланинов (см. раздел 9). Накопление субстратов-предшественников. При недостаточности фермента Е3 будут накапливаться вещество С, а также во многих случаях и предшествующие соединения. Увеличение субстратов-предшественников дефектного фермента - ведущее звено развития многих заболеваний:
Клинические проявления. Известно заболевание алкапгонурия, при котором нарушено окисление гомогентизиновой кислоты в тканях (гомогентизиновая кислота - промежуточный метаболит катаболизма тирозина). У таких больных наблюдают недостаточность фермента окисления гомогентизиновой кислоты - диоксигеназы гомогентизиновой кислоты, приводящей к развитию заболевания. В результате увеличиваются концентрация гомогентизиновой кислоты и выведение её с мочой. В присутствии кислорода гомогентизиновая кислота превращается в соединение чёрного цвета - алкаптон. Поэтому моча таких больных на воздухе окрашивается в чёрный цвет. Алкаптон также образуется и в биологических жидкостях, оседая в тканях, коже, сухожилиях, суставах. При значительных отложениях алкаптона в суставах нарушается их подвижность. Нарушение образования конечных продуктов и накопление субстратов предшественников.Отмечают заболевания, когда одновременно недостаток продукта и накопление исходного субстрата вызывают клинические проявления.
Клинические проявления. Например, у людей с болезнью Гирке (гликогеноз I типа) наблюдают снижение концентрации глюкозы в крови (гипогликемия) в перерывах между приёмами пищи. Это связано с нарушением распада гликогена в печени и выходом из неё глюкозы вследствие дефекта фермента глюкозо-6-фосфатфосфатазы (см. раздел 7). Одновременно у таких людей увеличиваются размеры печени (гепатомегалия) вследствие накопления в ней не используемого гликогена 49. Генные болезни в зависимости от типа их наследования. (рис. 4.7. стр 69, литвицкий!!!) В зависимости от функционального класса измененного полипептида (белки структурные, ферменты, рецепторы, трансмембранные переносчики и т.д.) делают попытки дифференцировать моногенные болезни на несколько классов. В настоящее время очевидно, что мутантные гены, кодирующие ферменты, приводят к развитию ферментопатий, наиболее часто встречающихся моногенных болезней. Для любого моногенного заболевания существенной характеристикой является тип наследования: аутосомно-доминантный, аутосомно-рецессивный, сцепленный с хромосомойX(доминантный и рецессивный), голандрический (сцепленный с хромосомойY) и митохондриальный (рис. 4-7). Клинические проявлениямоногенных болезней зависят от пенетрантности и экспрессивности гена. Пенетрантностьоценивается по проценту переносчиков, у которых обнаруживаются фенотипические проявления наследственного заболевания, вызванные экспрессией мутантного гена: если выявляется хотя бы один признак или симптом болезни, то считают, что фенотипические проявления гена имеются. Пенетрантность во многом зависит от воздействия факторов внешней среды. Например, у пациентов-гетерозигот с дефицитом глюкозо-6-фосфат-дегидрогеназы гемолиз эритроцитов происходит только под влиянием оксидантов. Следует помнить также, что при некоторых заболеваниях их симптомы появляются лишь в зрелом возрасте. Поэтому говорить о пенетрантности мутантного гена можно при достижении индивидуумом определенного возраста.
|
|||||||||
|
|
Последнее изменение этой страницы: 2021-03-09; просмотров: 84; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.15.153.69 (0.074 с.) |