
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Анализ продуктов изомеризацииСодержание книги
Поиск на нашем сайте
Анализ продуктов изомеризации циклогексана (метилциклопентана) проводят на хроматографе с детектором по теплопроводности. Для количественного расчета полученных хроматограмм предварительно проводят калибровку хроматографа и строят калибровочные графики. Готовят четыре искусственные смеси метилциклопентана с циклогексаном с концентрацией метилциклопентана 5, 10, 15 и 20% (масс.). Каждую смесь роматографии-руют не менее двух раз в условиях, приведенных выше. Находят среднее значение площадей пиков (Scp, мм2) для всех смесей и строят график зависимости: площадь пика — концентрация метилциклопентана (циклогексана) в смеси. Полученный график используют для нахождения концентрации метилциклопентана (циклогексана) в продуктах реакции. Контрольные вопросы 1. Назначение и технологические параметры процесса изомеризации. 2. Сырье установки изомеризации в промышленных условиях. 3. Реакции изомеризации на примере пентана, октана, гексадиена. Литература 1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 90-94. 2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с. 3. Паушкин Я.М., Вишнякова Т.П., белов П.С. Практикум по нефтехимическому синтезу. М.. Химия, 1965. 208 с. Лабораторная работа №4 ПИРОЛИЗ УГЛЕВОДОРОДОВ Теоретическая часть
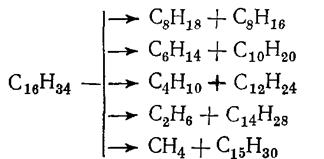
Углеводороды подвергаются наиболее глубоким химическим превращениям в процессах пиролиза, которые проводят при температурах выше 700°С. В таких условиях углеводороды подвергаются дегидрированию и расщеплению по углерод-углеродным связям с образованием газообразных, жидких и твердых продуктов. Например, первичным продуктом расщепления н-гексадекана могут быть следующие соединения, которые, в свою очередь, подвергаются дальнейшему распаду по аналогичной схеме.
Процессы распада усиливаются с повышением температуры и времени контакта, при этом в продуктах пиролиза накапливаются вещества, более стабильные в данных условиях. При пиролизе любого углеводородного сырья в интервале 750— 850 °С целевыми веществами в газообразных продуктах реакции являются этилен, пропилен и бутилен, а в жидких — ароматические углеводороды. При дальнейшем повышении температуры пиролиза в газообразных продуктах появляется заметное количество ацетилена, усиливается выделение водорода и образование кокса, так: как выше 850 °С заметно возрастают скорости разложения углеводородов на элементы и дегидроконденсации (уплотнения) ароматических углеводородов:
В условиях пиролиза все реакции углеводородов протекают через образование свободных радикалов, которые возникают в результате расщепления связей С — С иод воздействием высокой температуры:
Свободные радикалы, в свою очередь, инициируют радикально-цепные реакции превращения углеводородов:
Пиролиз углеводородов, как правило, проводят в присутствии паров воды, что значительно смягчает условия процесса, снижает долю вторичных реакций и препятствует образованию кокса. Расход водяного пара на пиролиз колеблется от 25 до 100% от массы исходного углеводородного сырья. Цель работы Изучение влияния температуры и объемной скорости подачи исходного сырья на выход и состав газообразных и жидких продуктов пиролиза углеводородов. Реактивы н-Октан (бензин, лигроин) Азот (из баллона) Хлорид кальция (безводный)
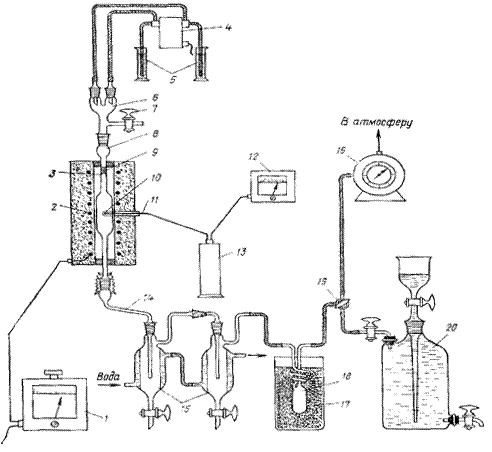
Методика проведения опыта Пиролиз н-октана проводят на установке, схема которой приведена на рисунке 4. Установка состоит из автотрансформатора 1 электропечи 2, выступов 8, дозатора 4, мерных цилиндров 5, переходников 6 и 14, крана 7, реактора 8, насадки 9, кармана для термопары 10, термопары //, милливольтметра 12, ледяных бань 13 и 17, приемников 15, газовых часов 16, ловушки 18, трехходового крана 19 и газометра 20. Перед началом опыта проверяют правильность сборки, надежность и герметичность всех соединений установки. Проверяют правильность работы электромеханического дозатора 4 для подачи в реактор 8 углеводорода и воды. Наливают в один из мерных цилиндров 5 углеводород, а в другой дистиллированную воду. Устанавливают на дозаторе заданные скорости подачи исходных веществ, отсоединяют линии подачи углеводорода и воды от переходника 6 и концы этих линий опускают в отдельные цилиндры емкостью 10 мл. Включают дозатор и по секундомеру определяют истинную скорость подачи исходных веществ; при необходимости проводят регулировку дозатора. По окончании проверки дозатор выключают, а линии подачи воды и углеводорода снова присоединяют к переходнику 6. Переключают трехходовой кран 19 на газовые часы 16 и через кран 7 продувают установку азотом со скоростью 100 мл/мин в течение 10 мин. После этого прекращают подачу азота, закрывают кран 7, включают электропечь 2 для обогрева реактора и приступают к выполнению опытов.
Рисунок 4. Установка для пиролиза углеводородов: 1 – автотрансформаторы, 2 – электропечь, 3 – выступы, 4 – дозаторы, 5 - мерные цилиндры, 6, 14 – переходники, 7 – кран, 8 – реактор, 9 – насадки, 10 - карман для термопары, 11 – термопара, 12 – милливольтметр, 13, 17 – ледяные бани, 15 – приемники, 16 – газовые часы, 18 – ловушки, 19 – трехходовой кран, 20 – газометр.
Заданием предусматривается выполнение работы по одному из следующих вариантов: 1) при постоянном массовом отношении углеводорода к воде, равном 2:1, варьируют объемную скорость подачи углеводорода (0,5, 1,0 и 2,0 ч-1) при двух уровнях температур (750 и 850 °С) — всего 6 опытов; 2) исследуют процесс методом планирования эксперимента при постоянном массовом отношении углеводорода к воде (2:1) и двух уровнях объемной скорости подачи углеводорода (1 и 2 ч -1) и температуры (750 и 850 °С) с постановкой 3 опытов в центре плана— всего 7 опытов. На один опыт расходуют 40 мл углеводородного сырья. При необходимости проверяют воспроизводимость опытов. В процессе проведения опыта наблюдают за отложением кокса на стенках реактора. При значительном его отложении перед началом нового опыта очищают стенки реактора. Для этого присоединяют к крану 7 линию азота и через установку пропускают азот со скоростью 100 мл/мин в течение 10 мин. Отсоединяют линию азота, но кран 7 не закрывают; присоединяют к приемнику 15 вместо ловушки 18 водоструйный насос и просасывают через установку воздух при 750—780°С до полного сгорания кокса. После этого отключают водоструйный насос, присоединяют к приемнику 15 ловушку 18, вновь продувают установку азотом в течение 10 мин и затем приступают к выполнению следующего опыта. Реактор представляет собой кварцевую трубку с расширенной реакционной зоной объемом около 30 мл (диаметр 20 мм). Он снабжен карманом 10 для термопары 11 и выступом 3 для удерживания кварцевой насадки 9. Высота слоя насадки 15 мм, размер частиц 4—5 мм. Заданную температуру в реакторе поддерживают с точностью ±5°С с помощью автотрансформатора / (или терморегулятора). По достижении в реакторе заданной температуры и стабилизации ее включают вначале подачу воды, а затем (через 5 мин) подачу углеводорода. Момент включения подачи углеводорода принимают за начало опыта. Продукты пиролиза через переходник 14 поступают в приемники 15, охлаждаемые водой, затем в ловушку 18, охлаждаемую ледяной баней 17, где конденсируются и собираются жидкие продукты пиролиза. Газы проходят через трехходовой кран 19, газовые часы 16 и выпускаются под тягу. Через 10 мин после начала опыта в газометр 20 отбирают 3 л газов пиролиза для анализа. По окончании опыта выключают вначале подачу углеводорода, а затем (через 5 мин) подачу воды и обогрев реактора. После каждого опыта фиксируют объем газов, прошедших через газовые часы, и объем газов, собранных в газометр. Определяют плотность и общую массу газов пиролиза, а также содержание в них водорода, этилена и пропилена. Жидкие продукты пиролиза из приемников и ловушки объединяют, отделяют от воды и сушат над безводным хлоридом кальция; находят их массу, перегоняют и анализируют. Составляют материальный баланс опыта. На основании полученных данных строит график зависимости выхода (в % масс.) газообразных и жидких продуктов пиролиза от объемной скорости подачи углеводородного сырья при двух температурах, а также выхода (в % масс.) этилена и пропилена. При исследовании процесса методом планирования эксперимента выводят регрессионные уравнения для выхода газообразных и жидких продуктов, этилена и пропилена. Обсуждают полученные зависимости и формулируют свои выводы по результатам проведенной работы. Анализ газов пиролиза
Газы пиролиза анализируют на хроматографе с детектором по теплопроводности в два приема. В одной пробе определяют только содержание водорода, в другой — содержание остальных компонентов газа. Анализ жидких продуктов пиролиза
Жидкие продукты пиролиза из всех опытов объединяют, взвешивают, добавляют к ним 1 г гидрохинона для предотвращения полимеризации непредельных и осторожно перегоняют из колбы Кляйзена. После отгонки фракции, выкипающей до 200 °С, перегонку прекращают. Определяют массу отогнанной фракции и анализируют ее на содержание непредельных и ароматических углеводородов. Остаток в колбеКляйзена относят к смолистым веществам.
Контрольные вопросы 1. Сущность процесса пиролиза углеводородов. 2. Сырье и применение продуктов пиролиза. 3. Основные и побочные реакции пиролиза.
Литература 1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 94-99. 2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с. 3. Паушкин Я.М., Вишнякова Т.П., белов П.С. Практикум по нефтехимическому синтезу. М.. Химия, 1965. 208 с.
Лабораторная работа №5 Тема: ОКИСЛЕНИЕ АММИАКА
Теоретические основы
Окисление аммиака до окиси азота является начальной стадией производства азотной кислоты. Наилучшим избирательным действием на процесс окисления аммиака до окиси азота обладает платина, которая при высоких температурах может давать выход до 99 %. При проведении процесса на платиновых катализаторах окисление аммиака начинается уже при 145°С с получением главным образом элементарного азота и закиси азота. Наибольший выход окиси азота 95-98 % достигается при температурах 850-950 ° С. При повышении температуры возрастают потери платины, что приводит к необходимости ограничить несколько верхний температурный предел процесса. Повышение температуры выше 1 000°С приводит к побочным процессам. Чрезмерное повышение температуры исходного газа сопровождается диссоциацией аммиака и окиси азота. Оптимальные температуры окисления аммиака на платине при атмосферном давлении находятся в интервале 800-840°С; для окисленных катализаторов оптимальные температуры несколько ниже и составляют 700-800 °С. В качестве окисляющего агента для процесса окисления аммиака используется в подавляющем большинстве кислород воздуха. Расход кислорода на окисление аммиака до окиси азота может быть определен согласно уравнению:
4NH3 +5O2 → 4NO+6H2О
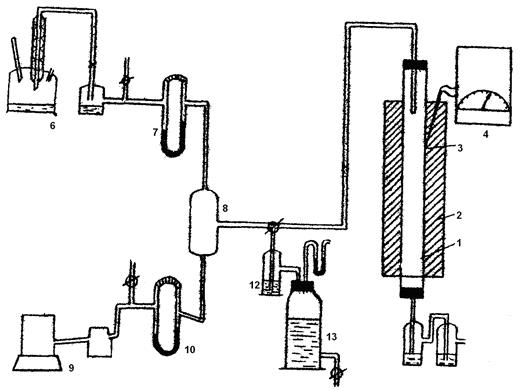
Таким образом, стехиометрическое мольное соотношение кислорода к аммиаку O2/NH3 составляет 1,25. Однако при таком соотношении выход окиси азота незначителен. Для его повышения необходим определенный избыток кислорода, от которого будет зависеть и концентрация аммиака в воздушно-аммиачной смеси. В производственных условиях содержание аммиака в воздушно-аммиачной смеси поддерживается в интервале 9,5-11,5 %, что соответствует отношению O2/NH3 - 1,7-2,0. Для окисных катализаторов, имеющих меньшую, чем платина активность, соотношение O2/NH3 должно быть более двух и соответственно концентрация аммиака в воздушно-аммиачной смеси должна поддерживаться в пределах 7,5-9,5 %. При изменении концентрации аммиака в исходной смеси следует учитывать, что воздушно-аммиачные смеси могут быть в определенном интервале взрывоопасны. Применяемые в промышленности аммиачно-воздушные смеси (9,5-11,8 %) практически не представляют опасности. Что касается аммиачно-кислородных смесей, то они воспламеняются со взрывом при любой концентрации аммиака в интервале температур 700-800 ° С. Реакция окисления аммиака до окиси азота протекает во внешне диффузионной области как на платине, так и на окисных катализаторах. Сама реакция протекает быстро, но общая скорость процесса вследствие внешнедиффузионного торможения может снижаться более чем на 90 %. Максимальной степени превращения аммиака в окись азота соответствует определенное время контакта. Практическое осуществление процесса окисления аммиака требует таких условий, которые обеспечивают максимальное протекание основной реакции и минимальной потери аммиака в виде молекулярного азота. С этой целью окисление аммиака проводят в присутствии активного, избирательно действующего катализатора, в сравнительно узком интервале температур, при строго определенных времени соприкосновения газа с катализатором и начальном составе аммиачно-воздушной смеси. Наиболее распространенным промышленным катализатором процесса является платина или ее сплавы с родием и палладием. Для обеспечения большой поверхности катализатора платиновые катализаторы обычно используются в виде сеток из проволоки диаметром 0,045-0,09 мм, причем свободное сечение сетки составляет 50-60 %. Достаточно полный контакт газа с катализатором достигается установкой в аппарате последовательно трех сеток. Из всех металлов платиновой группы наиболее оптимальными являются добавки родия. Сплавы платины с родием характеризуются не только более высокой каталитической активностью, чем чистая платина, но имеют лучшие прочностные показатели. Особенно это важно при работе в области высоких температур, так как у них точка плавления сплава выше, чем у платины. Применяются в промышленности также палладиевые катализаторы, несколько уступающие платинородиевым по механической прочности, но имеющие высокую каталитическую активность. Различные сплавы платины и палладия с добавками родия, серебра, иридия, кобальта, вольфрама и др. обеспечивают выход окиси азота 96-99 %. Из неплатиновых катализаторов окисления аммиака наиболее активными оказались катализаторы на основе оксидов железа и кобальта; активированные добавки хрома, марганца, висмута, никеля и др.; многие из этих катализаторов показали высокую активность. Так, на окисножелезном катализаторе, промотированном окислами висмута и марганца, степень конверсии достигает 94 %. Однако, для всех окисных катализаторов наблюдается потеря активности во времени. Для систем, работающих при атмосферном давлении, применяется двухступенчатое окисление аммиака: 1-ая ступень платинородиевая сетка, 2-ая ступень - слой таблетированного окисного (чаще железо-хромового) катализатора, толщиной 50-65 мм. Общий выход окиси азота в таких аппаратах 96,5 %, затраты на платину в этом случае сокращаются в 3 раза, снижаются и потери платины. Все окисные катализаторы относятся к типу осажденных и готовятся совместным осаждением гидроокисей из растворов серно- и азотнокислых, хлористых солей. Неплатиновые катализаторы на носителях практически не применяются вследствие малой активности. Согласно теории диффузионной кинетики окисления аммиака. Скорость процесса его окисления на платине определяется наиболее медленной стадией - диффузией аммиака к поверхности катализатора. Продолжительность реакции окисления аммиака на платине составляет 10 -4-10 -5 сек. Время контактирования может быть определено из уравнения:
где V св - свободный объем катализатора, м3 /сек; Vr - объемная скорость газа в условиях конверсии, м3 /сек.
Существует вполне определенное время контакта и соответственная ему скорость газового потока, обеспечивающие максимальный выход окиси азота. Для осуществления реакции необходимо соответствующее энергетическое состояние реагирующей системы, определяемое значениями энергии активации. Процесс каталитического окисления аммиака начинается со стадии активированной адсорбции кислорода на поверхности катализатора с образованием промежуточного соединения, затем происходит активированная адсорбция аммиака, требующая меньшей энергии активации; при этом образуется переходной комплекс с последующей перегруппировкой его в комплекс. Возможно образование других промежуточных соединений гидроксиламина NH2OH, нитроксила NHO, амида NH2, атомарного азота и др. и в конечном счете образование NO, N2, H2O. Из всех термодинамически возможных реакций в присутствии катализатора в первую очередь протекает та реакция, которая требует наименьшей энергии активации. В соответствии с адсорбционно-химической теорией катализа механизм каталитического окисления на платине можно представить следующим образом. Кислород и аммиак диффундируют из потока к поверхности катализатора. Находящиеся на поверхности платины атомы со свободными валентностями обеспечивают первоочередную активированную адсорбцию кислорода за счет возникновения электронной связи. Химическое взаимодействие платины с кислородом приводит к ослаблению атомарных связей в молекулах и образованию перекисного комплекса катализатор - кислород. Последующая активированная адсорбция аммиака дает в результате комплекс катализатор-кислород-аммиак и в конечном результате происходит перераспределение электронных связей и соединение атомов азота и водорода с кислородом. Адсорбированный кислород не входит в кристаллическую решетку платины и образует с ней непрочные связи. Молекулы аммиака ориентируются к кислороду атомами водорода (вследствие высокого сродства кислорода и водорода) с последующим образованием молекул окиси азота и воды. NO и Н2О свойственна малая адсорбционная способность; вследствие этого они десорбируются с поверхности катализатора, освобождая связи для вновь сорбируемых молекул кислорода. Механизм окисления аммиака на окисных катализаторах, по-видимому, не будет иметь принципиального отличия от такого на платине; однако, оптимальные условия процесса (температура, состав газа, время контактирования) в этом случае изменяются в зависимости от стадии, лимитирующей процесс в целом. Целью данной работы является испытание активности неплатиновых катализаторов на лабораторной модельной установке в зависимости от условий: температуры, объемной скорости газа, его начального состава, времени контактирования и т.д. Методика проведение опыта
Контактный аппарат представляет собой кварцевую или фарфоровую трубку с катализатором 1, установленную в вертикальной (или горизонтальной) трубчатой печи 2 с электрообогревом. Температура в контактном аппарате заменяется термопарой — 3 и регулируется автоматическим терморегулятором 4. В кварцевую трубку помещён неплатиновый гранулированный катализатор. Трубка имеет длину около 300 мм и внутренний диаметр 20-30 мм. Газ подается в контактный аппарат сверху через стеклянную трубку 5. Газовая смесь готовится следующим образом: аммиак из склянки 6 через реометр 7 подается для смешения в стеклянный или алюминиевый смеситель 8 с воздухом, подаваемый воздуходувкой 9 через реометр 10. Из смесителя газ подается в контактный аппарат и проходит в нем сверху вниз. Для поглощения окислов азота газ после контактного аппарата пропускается через поглотительную склянку 11 с раствором перекиси водорода и отводится далее в тягу. Опыт проводится в следующем порядке: расходы аммиака и воздуха, требуемые для получения смеси заданного состава, устанавливают по заранее градуированному реометру. Общая объемная скорость газовой смеси задается в пределах 200-1000 см3/мин. Состав газа устанавливают и контролируют путем анализа. Газовая смесь с определенной заданной скоростью подается в контактный аппарат, нагретый до заданной температуры. Температура задается в пределах 600-850 ° С. Во время опыта скорость газа и температура в контактном аппарате поддерживается строго постоянной. В течение опыта 2-3 раза, через каждые 10 минут, отбираются пробы газа для анализа содержания аммиака. Таким же образом проводятся опыты при других заданных условиях. Измеряемыми параметрами могут быть состав исходного газа, температура, время контактирования и состав исходного катализатора. Для каждого данного катализатора при измерении одного из факторов технологического режима остальные условия опыта должны быть строго постоянными. Время контактирования можно изменить либо путем изменения скорости газа, либо высоты слоя катализатора. Время контактирования рассчитывается исходя из объемной скорости газового потока и свободного объема контактной зоны VCB.
Рисунок 5 - Схема лабораторной установки окисления аммиака
1 – трубка с катализатором; 2 – трубчатая печь; 3 – термопара; 4 - терморегулятор; 5 – стеклянная трубка; 6, 11, 12 – склянки; 7, 10 – реометры; 8 – смеситель; 9 – воздуходувка; 13 – газометр
Исходная газовая смесь анализируется на содержание NH3 пропусканием газового потока при помощи аспиратора и крана через поглотительную склянку (дрексель), в которую предварительно заливают 50 см3 воды, 10 см3 0,1 н раствора НС1 и несколько капель метилового оранжевого. Поглотительная склянка соединена с аспиратором, создающим разряжение для протягивания газа через поглотительную склянку. Газ пропускается через поглотитель до тех пор, пока красная окраска раствора не перейдет в желтую. Количество воздуха в отобранной на анализ пробе аммиачно-воздушной смеси определяют по объему воды, вытекающей из аспиратора. Содержание аммиака в газе (в объемных %) вычисляется по формуле:
где а - объем NH3, см3, V - объем вытекающей воды из аспиратора, см3 Р1 - давление по барометру, мм.рт.ст., t - температура в помещении, ° С, Р2- упругость водяных паров при температуре, мм.рт.ст.
Анализ газа после контактирования проводится методом эвакуированных колб. В колбу заливают 5 см3 3 %-ого раствора перекиси водорода и, поместив колбу в матерчатый мешок (или обернув ее полотенцем), присоединяют с помощью резинового шланга к вакуумнасосу: колбу вакуумируют до остаточного давления 200-300 мм рт. ст., проверяют герметичность шлифа колбы; для этого колбу переворачивают горлом вниз и наблюдают, не происходит ли всасывания воздуха (если колба не герметична, то видны пузырьки воздуха, проходящие через раствор перекиси водорода). Если шлиф колбы не герметичен, то следует вынуть пробку со шлифом, протереть горло колбы насухо (фильтровальной бумагой), смазать его вакуумной смазкой, протереть пробку со шлифом в горловине и вновь вакуумировать. Далее колбу взвешивают на аналитических весах и засасывают в нее анализируемую газовую смесь, присоединяют колбу к газоотборнику. Перед взятием пробы сливают конденсат и продувают анализируемую газом трубку газоотборника. Отбор газа в колбу проводят осторожно, проворачивая трубку со шлифом до совмещения отверстий в шлифе с отверстием газоотборного отростка горловины колбы. При повороте колбы следят за уровнем жидкости в манометре, на реометре - воздуха, прикрывая и открывая вновь отверстия для газоотбора, с учетом того, чтобы не происходил переброс жидкости в реометр. Отбор газа производят до прекращения колебаний жидкости в манометре, т.е. выравнивая давление в колбе и установке. После взятия пробы газа, колбу взвешивают снова и определяют навеску газа по разности. Затем колбу встряхивают в течение 15-20 минут. При этом кислород воздуха, имеющийся в колбе, и перекись водорода окисляют окислы азота в азотную кислоту, которую далее оттитровывают раствором едкого натра. С этой целью содержимое вакуумированной колбы переливают в коническую колбу, куда добавляют и промывные воды от промывания вакуумированной колбы несколькими порциями дистиллированной воды. Перед титрованием необходимо разрушить избыток перекиси водорода непродолжительным кипячением; перед кипячением к анализируемому раствору добавляют отмеренное количество титрованного 0,1 н раствора едкого натра. Количество едкого натра берут по заданию преподавателя в избытке в зависимости от концентрации газа и его навески. Избыток едкого натра оттитровывают после кипячения и охлаждения пробы 0,1 н раствором НСl с метиловым оранжевым — в качестве индикатора. Количество едкого натра, израсходованное на нейтрализацию азотной кислоты, пересчитывают на окисленный аммиак. Расчет результатов анализа производится по формуле:
где C2 - содержание окисленного аммиака в конечном газе, % вес.; а - количество см3 0,1 н раствора NaOH, прилитое к анализируемому раствору; в - количество см3 ОД н раствора НС1, израсходованного на титрование избытка NaOH; (а-в)0,0017 - количество аммиака, окисленного до NO в пробе газа, г; G - вес пробы. Для вычисления степени окисления X пересчитывают содержание аммиак в исходном газе также на весовые проценты по формуле:
где С1 - концентрация NH3, % об.; 17 и 28 - соответственно молекулярные веса аммиака и воздуха.
Контрольные вопросы 1. Теоретические основы процесса окисления аммиака. Применение продуктов окисления. 2. Механизм окисления аммиака на окисных катализаторах. 3. Как определяется содержание аммиака в газе? 4. Анализ получаемого газа.
Литература 1. Исагулянц В.И., Егорова Г.М. Химия нефти. Руководство к лабораторным занятиям. 2-е изд. М., Химия, 1965, 517 с. 2. Лабораторный практикум по технологии основного органического синтеза/ Рейхсвельд В.О., Рубан В.Л., Саратов И.Е. М., Химия, 1966, 320 с. Лабораторная работа №6
|
||||
|
|
Последнее изменение этой страницы: 2020-12-09; просмотров: 233; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.119 (0.011 с.) |

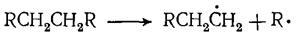

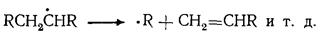
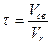
 (5.1)
(5.1)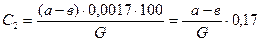 (5.2)
(5.2)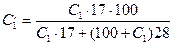 (5.3)
(5.3)


