Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Глава 1. Современные методы и проблемы диагностики наследственной патологииСтр 1 из 5Следующая ⇒
СОДЕРЖАНИЕ ВВЕДЕНИЕ ГЛАВА 1. СОВРЕМЕННЫЕ МЕТОДЫ И ПРОБЛЕМЫ ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ 1.1 Современные представления о наследственных заболеваниях Геномный импринтинг Болезни импринтинга ГЛАВА 2. ХАРАКТЕРИСТИКА СИНДРОМОВ ПРАДЕРА-ВИЛЛИ И АНГЕЛЬМАНА Цитогенетическая характеристика синдромов Прадера-Вилли и Ангельмана 2.2 Клинические проявления и методы диагностики синдромов Прадера-Вилли и Ангельмана Клинические проявления синдрома Прадера-Вилли Клинические проявления синдрома Ангельмана Связь синдромов Прадера-Вилли и Ангельмана ГЛАВА 3. СИНДРОМ ДИ ДЖОРДЖИ (ДИ ГЕОРГА) ВЫВОДЫ СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
ВВЕДЕНИЕ
Одной из наиболее актуальных проблем современной медицинской генетики является определение этиологии и патогенеза наследственных заболеваний. Цитогенетические и молекулярные исследования имеют высокую диагностическую информативность и ценность при решении данной проблемы, так как хромосомные аномалии встречаются с частотой от 4 до 34% при различных наследственных синдромах. В последние годы появились новые методы цитогенетических и молекулярно-цитогенетических исследований, значительно расширяющие диагностические возможности при заболеваниях, сопровождающихся различными хромосомными нарушениями. Данный обзор посвящен вопросам выбора необходимых цитогенетических или молекулярно-генетических анализов при различных формах генетических синдромов. В работе рассматриваются генетические заболевания, вызванные микроделециями хромосом. На примере наследственных заболеваний (синдромах Прадера-Вилли, Ангельмана, Ди Джорджи) будет изучено явление геномного импринтинга, влияние которого ученые изучают и сейчас, так как до конца механизм геномного импринтинга пока не известен. Будут рассмотрены возможные варианты медицинской помощи при данных заболеваниях и риски дальнейших проявлений этих заболеваний в семьях, где встречаются такие патологии. Изучение этой проблемы на сегодня является актуальной. Целью работы является: изучение цитогенетических и клинических проявлений микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи.
ГЛАВА 1. СОВРЕМЕННЫЕ МЕТОДЫ И ПРОБЛЕМЫ ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
Геномный импринтинг
В середине XIX в. Грегор Мендель в своих опытах по скрещиванию гороха сделал наблюдение, которое впоследствии стало настоящей аксиомой для генетиков. Он обнаружил, что, если скрестить гомозиготное растение, имеющее гладкие семена, и гомозиготное растение с морщинистыми семенами, в потомстве все растения будут одинаковыми и дадут только гладкие семена. Этот результат не зависел от того, у какого из растении, взятых для скрещивания, - мужского или женского - семена были гладкими. Так Мендель открыл принцип эквивалентности реципрокных скрещиваний: у потомства ген действует одинаково независимо от того, от кого из родителей он унаследован. Трудно переоценить значение этого наблюдения Менделя в истории и практике генетики. Было установлено, что такой закономерности подчиняется большое число наследственных признаков - не только у гороха, но и у многих других организмов [3]. Исключения из правила идентичности гибридов при реципрокных скрещиваниях (т. е. скрещиваниях между двумя формами, когда каждая из них в одном случае берется в качестве матери, а в другом - в качестве отца) в действительности известны давно, однако, как правило, их можно было отнести к одному из двух классов. Первый составляют признаки, которые определяются генами, расположенными в половых хромосомах: у самок млекопитающих в ядрах клеток имеется по две Х-хромосомы, у самцов - по одной Х- и одной У-хромосоме. Например, цветовая слепота и гемофилия связаны с генами X-хромосомы. Наследование этих сцепленных с полом признаков подчиняется вполне определенным правилам, согласно которым гибриды в реципрокных скрещиваниях не обязательно идентичны. Рассмотрим пример: у отца-дальтоника и нормальной по этому признаку матери ни один из сыновей не будет дальтоником. Если мать страдает дальтонизмом, а отец нет, то все сыновья окажутся дальтониками. В обоих случаях дочери будут нести ген, обусловливающий дальтонизм, но иметь нормальное зрение. Наследование и проявление признаков, сцепленных с полом, зависят от пола потомка, но не связаны непосредственно с полом того родителя, от которого унаследован признак.
Второй класс неэквивалентных реципрокных скрещиваний включает признаки, определяемые внеядерными генами. Некоторые клеточные органеллы - митохондрии в клетках животных, митохондрии и хлоропласты в клетках растений - обладают своей собственной генетической информацией. Эти органеллы передаются из поколения в поколение с цитоплазмой яйцеклеток и поэтому наследуются исключительно по материнской линии. Таков характер наследования цвета листьев у некоторых растений, а также заболевания человека, известного под названием митохондриальной энцефаломиопатии. При митохондриальном наследовании зигота, образующаяся в результате слияния половых клеток, получает митохондрии и содержащуюся в них мтДНК только через яйцеклетку [3]. Недавно генетики и эмбриологи описали третье исключение- это геномный импринтинг, когда оба родителя передают потомкам совершенно идентичные гены, но эти гены несут специфический отпечаток пола родителей, т.е. отцовские и материнские гены активированы или супрессированы во время гаметогенеза по-разному. Таким образом, в некоторых случаях важно, от кого из родителей унаследован ген [1]. Суть геномного импринтинга заключается в том, что гены, передаваемые потомству, несут специфический «отпечаток» пола родителя, т.е. отцовские и материнские гены маркированы по-разному; причем эти «отпечатки» временные и могут быть «стерты». Вследствие геномного импринтинга потомки, получившие маркированные гены от матери, отличаются от тех, которые унаследовали такие гены от отца. Другими словами, в некоторых случаях важно, от которого из родителей унаследован ген [1, 3]. Многие исследователи пытались установить молекулярную природу геномного импринтинга, обеспечивающие его механизмы, а также число и функции маркируемых генов. Благодаря этому сделано несколько замечательных открытий, которые расширяют понимание ряда раковых и наследственных заболеваний, а также некоторых других патологий. Изучение геномного импринтинга, возможно, откроет что-то новое и в наследовании признаков, которые вполне удовлетворительно объясняются в рамках классической менделевской генетики [3]. Термин «импринтинг»(imprint - отпечаток) впервые предложил в 1960 г. Х. Кроуз из Колумбийского университета США для описания селективной элиминации отцовских хромосом у насекомых. Геномный импринтингназывают эпигенетическим явлением,подчеркивая этим, что наследуются изменения генной активности, обусловленные родительским происхождением хромосом или их фрагментов, а не структурные перестройки генетического материала (мутации). Таким образом, в некоторых участках генома, подверженных геномному импринтингу, экспрессируется только один отцовский или материнский аллель, т.е. наблюдается моноаллельная экспрессия импринтированных генов (генов, которые дифференциально экспрессируются в зависимости от отцовского или материнского происхождения) в отличие от обычной диаллельной. Причем, если импринтирован материнский ген, то экспрессируется отцовский аллель и наоборот. Наличие такого способа регуляции работы генов свидетельствует о неэквивалентном вкладе родителей в функционирование генома потомков, а фенотипические признаки, контролируемые импринтированными локусами, могут появляться в результате не только мутаций генов, но и нарушения эпигенетической программы регуляции генной экспрессии [1].
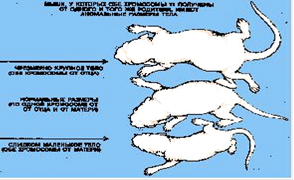
Геномный импринтинг занимает особое место среди специфических механизмов регуляции активности генов на ранних стадиях развития, приводя к различиям в экспрессии гомологичных материнских и отцовских аллелей. Первоначальный «отпечаток», созданный в половых клетках, служит основанием для дальнейших модификаций в результате взаимодействий между родительскими геномами и цитоплазматическими факторами яйцеклетки во время формирования пронуклеуса (автономное существование яйцеклетки и сперматозоида в зиготе). Последующие эпигенетические модификации могут привести к тому, что изменения в экспрессии генов будут стабильно передаваться в процессе развития клеточных поколений. Геномный импринтинг, например, может изменять дозу генов, контролирующих рост эмбриона, клеточную пролиферацию и дифференцировку [1]. Изучение геномного импринтинга у млекопитающих началось в начале 80-х годов XX в. после опытов на мышах, проведенных Дж. Мак Гратом, Д. Солтером и М. Сурани (рис. 1). Авторы разработали тонкий микрохирургический метод переноса клеточных ядер мышиных эмбрионов в стадии пронуклеусов и показали, что наследование хромосомных наборов только от одного из родителей приводит к нарушению процесса развития. Оказалось, что отцовский генетический вклад важен для развития плаценты, а материнский вклад необходим для развития тела эмбриона. Далее было показано, что наследование части индивидуальных хромосом или целой хромосомы только от одного из родителей может также приводить к аномальному фенотипу [3].
Рис. 1 Геномный импринтинг у мышей
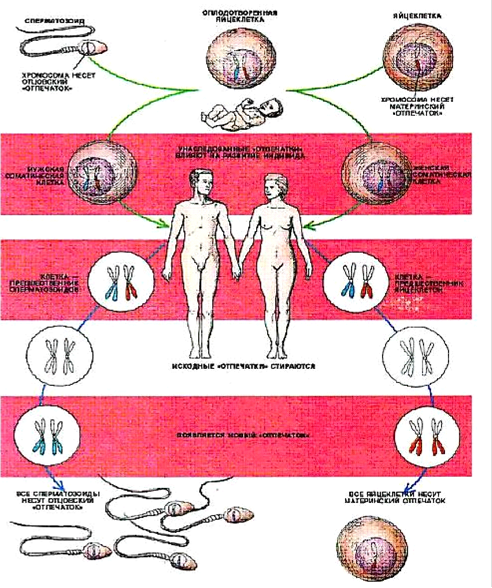
Рис. 2 Геномный импринтинг у человека Таким образом, геномный импринтинг состоит в том, что хромосомы половых клеток (сперматозоидов или яйцеклеток) индивида приобретают «отпечаток» его пола (рис. 2). Потомство получает один набор хромосом с отцовской маркировкой некоторых генов, а другой - с материнской. При образовании у потомка половых клеток прежний «отпечаток» стирается и эти гены маркируются в соответствии с полом данной особи.
Болезни импринтинга
Проявления геномного импринтинга удивительным образом связаны с некоторыми заболеваниями человека. Неожиданно оказалось, что в природе уже существуют параллели тем экспериментальным состояниям, которые исследовали у мышей. Недавно Р. Николлс и его коллеги из Медицинской школы Гарвардского университета установили, что у многих больных с синдромов Прадера-Вилли обе хромосомы 15 унаследованы от матери [3].
Р. Николлс, Дж. Кнолл (тоже сотрудник Гарвардского университета) и Ч. Уильямс из Флоридского университета обнаружили связанную с геномным импринтингом закономерность у больных с синдромом Ангельмана. У таких больных нередко имеют место частичные делеции унаследованной от матери хромосомы 15, в результате чего полностью функциональна только отцовская хромосома 15. Эти два заболевания, хотя и столь разные по клинической картине, могут быть связаны с различиями в импринтинге одних и тех же генов одной и той же хромосомы. Однако в отличие от ненормально крупных и мелких мышей, синдромы Прадера-Вилли и Ангельмана не удается представить просто как две стороны одной медали, т. е. объяснить избытком либо недостатком продуктов одних и тех же генов. Исследования Николлса и его коллег показали, что бывает очень трудно предсказать, каким образом конкретные признаки зависят от процесса геномного импринтинга. Полезно было бы изучить на этот предмет многие наследственные заболевания человека; не исключено, что найдутся указания на влияние геномного импринтинга [3]. Примером импринтинга целого генома у человека является истинный пузырный занос,который возникает при оплодотворении яйцеклетки, лишенной материнских хромосом, двумя сперматозоидами. Несмотря на наличие полноценного диплоидного набора, ранний эмбриогенез таких зигот протекает аномально: ткани собственно эмбриона вообще не формируются, однако бурно разрастается трофобласт. В случае двойного набора материнских хромосом развивается тератома - эмбриональная опухоль. Следовательно, у человека, как и у мыши, на ранних стадиях развития геном отца преимущественно обеспечивает развитие провизорных органов, а геном матери - эмбриональных структур. Только материнский или только отцовский геномы не в состоянии обеспечить нормальное развитие эмбриона [1, 3]. На организменном уровне эффект импринтинга обнаружен в связи с наличием в хромосомном наборе фрагментов или целых хромосом одного (материнского или отцовского) происхождения - так называемая однородытельская дисомия(ОРД), т.е. наблюдается качественный, а не количественный хромосомный дисбаланс. Известны два основных механизма образования ОРД у человека: коррекция трисомии до дисомии (гетеродисомия), происходящая в 1-м мейотическом делении, и коррекция моносомии до дисомии (изодисомия) - во 2-м мейотическом делении. Феноменология импринтинга значительно лучше изучена у мыши, чем у человека, и поскольку известна гомология между хромосомами человека и мыши (примерно по 700 локусам), можно использовать данные по импринтингу, полученные на мышах, для целенаправленного поиска импринтинга по определенным локусам у человека.
Импринтированные гены и их транскрипты обнаружены на многих хромосомах человека - 1, 5, 6, 7, 11, 13, 15, 19, 20и X. На хромосоме 7 мыши и хромосомах 1 и 15человека найдены два больших кластера ортологичных импринтированных генов, т.е. эволюционно консервативных по статусу импринтинга. Идентифицированы гены с полиморфным импринтингом, т.е. с сочетанием моноаллельной экспрессии в одних тканях и диаллельной - в других. По-видимому, такая тканеспецифическая эпигенетическая модификация некоторых генов может быть одним из механизмов, обеспечивающих дифференциальную экспрессию генов клеток разных тканей в ходе развития [1]. В последние годы с помощью молекулярно-генетических методов феномен геномного импринтинга наблюдают и при мультифакториальных заболеваниях. Например, четко выраженный отцовский импринтинг обнаружен при атопическом дерматите, материнский - при бронхиальной астме и атопии у детей. При инсулинзависимом сахарном диабете выявлена более высокая вероятность отцовского импринтинга. Ген инсулина у человека расположен в кластере импринтированных генов 11р15и гомологичен локусам в мышином геноме, подверженным импринтингу. Кроме того, обнаружена ОРД отцовского происхождения у детей с неонатальным сахарным диабетом. Примеров заболеваний, в основе этиологии которых лежит нарушение функции импринтированных участков генома, довольно много, поэтому можно говорить об особом классе заболеваний человека - «болезнях импринтинга»,которых насчитывается уже более 30. Основные из них приведены в табл. 1.
Таблица1Предполагаемые «болезни импринтинга» у человека
наследственность заболевание импринтинг микроделяционный синдром Рис. 4. Девочка с синдромом Прадера-Вилли Рис. 5. Возможные механизмы развития синдромов Прадера - Вилли и Ангельмана. В заключение следует еще раз подчеркнуть, что более широкое комплексное применение цитогенетических, молекулярно-цитогенетических методов лабораторного анализа в сочетании с грамотным клиническим обследованием позволит решить проблему диагностики изолированной умственной отсталости или умственной отсталости при наследственных болезнях и синдромах у детей. Это в свою очередь поможет более эффективно осуществлять помощь членам наследственно отягощенной семьи в решении вопросов прогноза потомства и снизить частоту рождения детей с генетическими патологиями.
ГЛАВА 3. СИНДРОМ ДИ ДЖОРДЖИ (ДИ ГЕОРГА) Синдром Ди Джорджи (Ди Георга, СДД) - изолированный Т-клеточный иммунодефицит. Характеризуется триадой ведущих клинических проявлений: гипоплазия тимуса и/или паращитовидных желез и врожденным пороком сердца. Впервые описан в 1965 году. Поражаются чаще девочки [9, 10, 11]. Наследуется по аутосомно - рецессивному типу [11]. Установлена генетическая характеристика этого врожденного порока развития, в части случаев это аутосомный генетический дефект делеции 22q11.2. В основе СДД лежит порок развития третьего-четвертого глоточных карманов, возникающий между шестой и десятой неделями гестации, приводящий к агенезии или дисгенезии паращитовидных желез и тимуса. Вовлечение первого и второго жаберных карманов приводит к пороку развития лицевых структур, а заинтересованность пятого кармана проявляется широким спектром врожденных пороков сердца с частым вовлечением дуги аорты [9]. Клиническая характеристика: у большинства больных отмечаются диспластические черты лица. Наиболее характерны диспластичные ушные раковины, гипертелоризм, широкая переносица, "рыбий рот", антимонголоидный разрез глаз. У части детей наблюдаются и более грубые аномалии, такие как микрогнатия и незаращение твердого и мягкого неба. Гипокальциемия различной степени тяжести и отсутствие тени вилочковой железы при рентгенографии грудной клетки относятся к частым проявлениям. Гипокальциемические судороги обычно возникают с первых дней жизни. У всех больных отмечается задержка умственного развития. Врожденные пороки сердца и магистральных сосудов также относятся к наиболее характерным и тяжелым признакам заболевания [9]. Тимус и паращитовидные железы образуются из третьего и частично четвертого карманов. Паращитовидные железы участвуют в регуляции уровня кальция в крови. Отсутствие данных желез приводит к судорогам вследствие гипокальциемии. Появление судорог у младенца в течение 48 часов после рождения свидетельствует о том, что у ребенка врожденная аплазия тимуса и дефицит Т-клеток. С данным синдромом связаны некоторые другие пороки развития, в том числе расщелина нёба (волчья пасть), щели в носу, низкопосаженные уши и неправильное формирование крупных кровеносных сосудов и сердца. Дети с пороками развития сердечно-сосудистой системы, как правило, умирают. Одно время считали, что этот синдром вызван нарушением нормального эмбрионального развития, которое не обусловлено наследственностью, поскольку не наблюдается семейной предрасположенности к заболеванию и оба пола поражаются одинаково. Впоследствии, однако, было установлено, что многие случаи заболевания связаны со структурными дефектами в 22-й хромосоме [9]. У детей, страдающих синдромом Ди Джорджи, либо совсем нет тимуса, либо он недоразвит, что встречается чаще. Такие дети чрезвычайно восприимчивы к вирусным, бактериальным и грибковым инфекциям и склонны к развитию таких опасных для жизни заболеваний, как цитомегаловирусная инфекция. Иногда в крови ребенка присутствует некоторое количество Т-клеток; такие дети способны сопротивляться инфекциям, при этом с возрастом число Т-клеток постепенно увеличивается [11]. Одна из форм нарушения иммунитета. Проявляется с рождения. Основные симптомы - тетания, обусловленная гипопаратиреозом; повышенная склонность к инфекциям, особенно вирусным и грибковым; аномалии развития костной системы, полости рта, сосудов, внутренних органов. Из инфекционных болезней чаще возникают бронхиты, пневмонии, диарея. Дети отстают и физическом развитии. Болезнь прогрессирует и без лечения к двум годам наступает летальный исход. При исследовании крови определяется низкий уровень, содержании кальции и высокий уровень фосфора, указывающие на паратиреендную недостаточность. Число лимфоцитов в периферической крови обычно нормальное, уровень иммуноглобулинов основных классов также в пределах нормы, но специфический иммунный ответ на введенный антиген снижен. Снижены реакции клеточного типа. Не развиваются и ослаблены реакции замедленной ин нечувствительности. Стимулированный лимфоузел обнаруживает избыточное число фолликулов и плазматических клеток. Наблюдаются уменьшение числа лимфоцитов в паракортикальных тимусзависимых зонах и белой пульпе селезенки, аплазия тимуса, гипоплазия паратиреоидных желез или аплазия их. Аплазия тимуса ведет к нарушению синтеза полноценных иммуноглобулинов. [12]. Иммунологический спектр: количественные показатели Т-клеток варьируют от нормы до глубокой депрессии. Характерна диссоциация между сниженными уровнями Т- и NK-клеток и повышенным содержанием В-лимфоцитов. Характерны нормальные или повышенные уровни антител [9]. Показатели гуморального иммунитета у больных с синдромом Ди Джорджа полностью нормальны. Концентрация иммуноглобулинов всех классов нормальная. У некоторых больных повышена концентрация lgE, что, возможно, связано с отсутствием Т-супрессоров. У части больных не удается получить антитела в ответ на иммунизацию [11]. 1. Обязательные лабораторные исследования: Анализ крови клинический 4 Тромбоциты 2 Определение группы крови и резус фактора 1 Анализ мочи 2 Определение уровня кальция в крови 2 Бактериологические исследования содержимого из очагов с определением чувствительности к антибиотикам 1 Общий белок и белковые фракции 1 Трансаминазы АсАТ, АлАТ 1 Маркеры вирусов гепатита В и С в крови 1 - С-реактивный белок 1 Сывороточные иммуноглобулины А, М, G 1 Субпопуляции лимфоцитов 2 Концентрация общего IgE в сыворотке 1 . Дополнительные лабораторные исследования: Определение уровня паратгормона в плазме 1 Вирусные маркеры (антитела к вирусу Эпштейна-Барр) 1 . Обязательные инструментальные исследования: Рентгенография органов грудной клетки УЗИ сердца ЭКГ . Дополнительные инструментальные исследования: УЗИ органов брюшной полости - 1. . Консультации специалистов (по показаниям): Эндокринолога - 1; Кардиохирурга - 1; Отоларинголога - 1; Окулиста - 1. [7] Характеристика лечебных мероприятий: иммунная недостаточность, за редким исключением, не определяет прогноз и спектр ведущих клинических проявлений при СДД. В большинстве случаев, если пациент переживает 6-месячный возраст, наблюдается постепенное спонтанное восстановление Т-клеточного иммунитета. Коррекция Т-клеточных нарушений при СДД может быть достигнута трансплантацией фетального тимуса. При наличии тяжелых пороков, в основном определяющих прогноз для жизни, пересадка тимуса считается недостаточно обоснованной. Лечение пороков сердца ведется по стандартам, принятым в кардиологии, а недостаточности паращитовидных желез - по стандартам эндокринологических отделений [9]. Для лечения синдрома Ди Джорджи с успехом применяют пересадку костного мозга. Кроме того, достигнуты положительные результаты при пересадке эмбрионального тимуса больным с тяжелой степенью поражения [11]. Лечение: трансплантация или имплантация ткани тимуса. Введение тимозина (экспериментально). Противосудорожная терапия, введение препаратов кальция [11]. Длительность стационарного лечения определяется характером и тяжестью клинических проявлений и инфекционных осложнений и варьирует от 3 недель до 3 месяцев [9]. Требования к результатам лечения - купирование клинических проявлений (осложнений) СДД [9, 10].
ВЫВОДЫ В представленной работе были рассмотрены микроделяционные синдромы: Прадера-Вилли, Ангнльма и Ди Джорджи. Описаны их клинические и генетические проявления, возможные риски. И представлены медицинские методы купирования их проявлений, улучшения состояния больных. На сегодняшний день синдромы Прадера-Вилли и Ангельмана служат общепринятой моделью для изучения новых в клинической генетике и сложных явлений - геномного импринтинга и унипарентальной дисомии. Установлено, что синдром Прадера-Вилли может быть обусловлен двумя основными механизмами. Первый из них - микроделеция хромосомы 15 (15q11.2-q13), которая всегда отцовского происхождения. Второй - материнская изодисомия, т.е. когда обе хромосомы 15 получены от матери. Развитие синдрома Ангельмана, наоборот, связано с микроделецией того же участка хромосомы 15, но материнского происхождения, или отцовской изодисомией. Большинство (около 70%) случаев синдрома Прадера - Вилли обусловлено микроделецией, остальные - дисомией. При этом обращает на себя внимание отсутствие клинических различий между больными с микроделецией и изодисомией. Для синдрома Ди Джорджи установлено, что он вызывается аутосомным генетическим дефектом делеции 22q11.2. Цитогенетические проявления данного синдрома на сегодня еще не достаточно изучены. Учеными ведется активная работа по решению этой проблемы.
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ 1. Гинтер Е. К. Медицинская генетика: Учебник. - М.: Медицина, 2003. - 448с. . Ридли М. Геном. Автобиография вида в 23 главах. - М.: Эксмо, 2008. - 432с. . Денисенкова Е. В., Петрин А. Н., Байков А. Д. Цитогенетические и молекулярно-цитогенетические аспекты диагностики заболеваний, сопровождающихся умственной отсталостью, у детей // Издательство «Медиа Сфера», 2005. Режим доступа: http://www.mediasphera.ru/journals/detail/206/2988/ . Сапиенца К. Геномный импринтинг // В мире науки. (Scientific American. Издание на русском языке). - 1990. - №12. - стр.14-20. Режим доступа: http://www.evolbiol.ru/gen.html . Дуліпа О. Г. Синдром Прадера-Віллі (Prader-Willi Syndrome). Режим доступа: http://www.ibis-birthdefects.org/start/... prader.htm . Казанцева Л. З., Новиков П. В., Семячкина А. Н., Николаева Е. А., Курбатов М. Б., Добрыкина Э. В. Московский НИИ педиатрии и детской хирургии Минздрава РФ. Синдром Прадера - Вилли у детей: новое в этиологии, патогенезе и лечении. Режим доступа: http://nature.web.ru/db/msg.html?mid=1174772&uri=index2.html . Миронов М. Б., Мухин К.Ю., Кузина Н.Ю., Боровиков К.С., Гоева И. А., Красильщикова Т. М., Коркин А. В., Петрухин А. С. Синдром Ангельмана. Клинический случай. Режим доступа: http://www.epileptologist.ru/pub/Angelman.html . Белозеров Ю. В., Леонтьева И. В., Школьникова М. А., Страхова О. С., Себелева И. А., Макаров Л. М., Давыдкин В. В., Динов Б. А. Наследственные болезни сердца у детей // Мир науки и культуры, 2000-2009. Режим доступа: http://nature.web.ru/db/msg.html?mid=1166264&s=111400060 . Соколов В. Н. Гипопаратериоз // Наука и практика, 2009. Режим доступа: http://www.chtfoms.ru/index.php?option=com_content&task=view&id=26467&Itemid=24 . Ярцев М. Н., Яковлева К.П., Плахтиенко М. В. ГНЦ Института иммунологии ФМБА России, Москва Иммунодефицитные состояния у детей // Издательство Media Medica, 2000. Режим доступа: http://old.consilium-medicum.com/media/pediatr/06_01/4.shtml . Вилочковой железы аплазия врожденная (Ди Джорджа синдром). Режим доступа: http://nasledbolezn.org.ua/obmen-veshestv/didjordja-sindrom.html СОДЕРЖАНИЕ ВВЕДЕНИЕ ГЛАВА 1. СОВРЕМЕННЫЕ МЕТОДЫ И ПРОБЛЕМЫ ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ 1.1 Современные представления о наследственных заболеваниях Геномный импринтинг Болезни импринтинга ГЛАВА 2. ХАРАКТЕРИСТИКА СИНДРОМОВ ПРАДЕРА-ВИЛЛИ И АНГЕЛЬМАНА Цитогенетическая характеристика синдромов Прадера-Вилли и Ангельмана 2.2 Клинические проявления и методы диагностики синдромов Прадера-Вилли и Ангельмана Клинические проявления синдрома Прадера-Вилли Клинические проявления синдрома Ангельмана Связь синдромов Прадера-Вилли и Ангельмана ГЛАВА 3. СИНДРОМ ДИ ДЖОРДЖИ (ДИ ГЕОРГА) ВЫВОДЫ СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
ВВЕДЕНИЕ
Одной из наиболее актуальных проблем современной медицинской генетики является определение этиологии и патогенеза наследственных заболеваний. Цитогенетические и молекулярные исследования имеют высокую диагностическую информативность и ценность при решении данной проблемы, так как хромосомные аномалии встречаются с частотой от 4 до 34% при различных наследственных синдромах. В последние годы появились новые методы цитогенетических и молекулярно-цитогенетических исследований, значительно расширяющие диагностические возможности при заболеваниях, сопровождающихся различными хромосомными нарушениями. Данный обзор посвящен вопросам выбора необходимых цитогенетических или молекулярно-генетических анализов при различных формах генетических синдромов. В работе рассматриваются генетические заболевания, вызванные микроделециями хромосом. На примере наследственных заболеваний (синдромах Прадера-Вилли, Ангельмана, Ди Джорджи) будет изучено явление геномного импринтинга, влияние которого ученые изучают и сейчас, так как до конца механизм геномного импринтинга пока не известен. Будут рассмотрены возможные варианты медицинской помощи при данных заболеваниях и риски дальнейших проявлений этих заболеваний в семьях, где встречаются такие патологии. Изучение этой проблемы на сегодня является актуальной. Целью работы является: изучение цитогенетических и клинических проявлений микроделяционных синдромов Прадера-Вилли, Ангельмана и Ди Джорджи.
ГЛАВА 1. СОВРЕМЕННЫЕ МЕТОДЫ И ПРОБЛЕМЫ ДИАГНОСТИКИ НАСЛЕДСТВЕННОЙ ПАТОЛОГИИ
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2020-03-26; просмотров: 117; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.219.213.196 (0.099 с.) |