Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Клинические проявления синдрома Прадера-ВиллиСодержание книги Поиск на нашем сайте
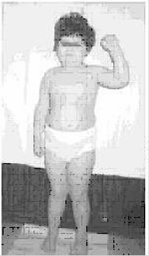
Синдром Прадера-Вилли (синдром Прадера-Лабхарта-Вилли-Фанкони, синдром НННО) описан в 1956 году является наиболее частой причиной генетически обусловленного тяжело протекающего ожирения у детей старше 1 года жизни и у взрослых. Встречается с одинаковой частотой среди обоих полов, регистрируется у лиц разных национальностей и рас. Популяционная частота составляет от 1:10 000 до 1:15 000 [5, 6]. Клиническая диагностика СПВ на 1-м году жизни затруднительна в силу отсутствия специфической симптоматики при рождении и быстрой спонтанной ликвидации ранних признаков заболевания после первых месяцев жизни. К этим ранним проявлениям синдрома относятся: вялое шевеление плода, слабость сосательного рефлекса вскоре после рождения, развитие мышечной гипотонии в первые месяцы жизни. Уже на 1-м году жизни могут выявляться различные дизморфии лица и конечностей, гипогонадизм и/или гипогенитализм. Наличие у ребенка первых месяцев жизни указанных симптомов нередко приводит врача к ошибочной диагностике пре- и перинатального поражения ЦНС, инфекционного процесса, так как часты нарушения терморегуляции; различных вариантов наследственных миопатий, врожденного гипотиреоза и других болезней. В существенной степени диагностику СПВ на 1-м году жизни затрудняют спонтанное восстановление сосательного рефлекса в первые месяцы жизни и повышение мышечного тонуса. Вместе с тем, развитие дефицитных заболеваний и необоснованное лечение по поводу другой предполагаемой патологии могут сказаться на последующем онтогенезе ребенка, в частности, его интеллектуальном развитии. Этот факт, несомненно, определяет актуальность ранней диагностики СПВ [5]. В большинстве случаев клиническая диагностика СПВ осуществляется после годовалого возраста ребенка, когда развивается 2-я фаза заболевания, характеризующаяся появлением у больного постоянного чувства голода. У ребенка постепенно и стойко формируется поведение «постоянного поиска пищи». Быстро развивается и прогрессирует ожирение, замедляются процессы роста, появляются умеренные признаки нарушения интеллекта, в дальнейшем нарушаются сроки полового развития и порядок появления вторичных половых признаков. Часто присутствует дневная сонливость, нередко по типу нарколепсии, а также ночные апноэ. В этой связи больные СПВ на протяжении своей жизни подвержены синдрому внезапной смерти. В патогенезе развития 2-й фазы заболевания основную роль играет гипоталамическая дисфункция, в том числе дефицит СТГ (гормона роста). Средняя окончательная длина тела без лечения СТГ у мальчиков составляет 155 см, у девочек - 147 см. Диагностика СПВ до развития 2-й фазы заболевания весьма актуальна. Она создает предпосылки для своевременного формирования у ребенка правильного поведения, в частности, пищевого. Коррекция дефицита СТГ, начатая до 18-месячного возраста ребенка, способствует формированию у него правильного телосложения, препятствует избыточному жироотложению, существенно улучшает развитие мышечной моторики. Последнее время в литературе ведется полемика о целесообразности длительной терапии соматотропином при СПВ. Возможности ранней диагностики СПВ в существенной степени повышаются при использовании диагностических методов, в основе которых лежит балльная оценка присутствующих у больного больших и малых признаков. К большим признакам (каждому присваивается 1 балл) относятся следующие: • характерные лицевые симптомы (долихоцефалия с уменьшением битемпорального диаметра, миндалевидный разрез глаз, небольшой рот с опущенными вниз углами и тонкой верхней губой, страбизм); • задержка нервно-психического развития до 6 лет, умеренное снижение интеллекта и проблемы обучения в школьном возрасте; • проблемы при кормлении в первые месяцы жизни с последующей нормализацией сосания в течение грудного периода; • изменения со стороны половой сферы; • мышечная гипотония центрального генеза в раннем детстве; • прогрессирующее ожирение в возрасте от 1 года до 6 лет. К малым признакам (каждому присваивается 0,5 балла) относятся следующие: • снижение двигательной активности плода и инфантильная летаргия; • нарушения рефракции; • снижение пигментации кожи и волос в сравнении с родителями; • следы «потертостей» кожных покровов; • поперечная ладонная складка; • низкорослость к 15 годам с учетом длины тела других членов семьи; • нарушения сна и апноэ во время сна; • маленькие стопы и/или кисти; • дефекты артикуляции и речи; • густая, вязкая слюна; • поведенческие нарушения [2, 5, 6]. Клинический симптомокомплекс Синдрома Прадера-Вилли включает сахарный диабет или нарушение толерантности к глюкозе. Наличие таких важных диагностических симптомов, как мышечная гипотония (Hypotonia), умственная отсталость (Hypomentia), гипогонадизм (Hypogonadism) и ожирение (Obesity), послужило основанием для одного из наименований синдрома - НННО. Тяжелая мышечная гипотония является наиболее ранним симптомом заболевания, возникает уже во внутриутробный период, что объясняет снижение подвижности плода. В ранний постнатальный период имеют место снижение сухожильных, глотательного и сосательного рефлексов, затрудняющих кормление, дыхательные нарушения, малоподвижность, задержка развития двигательных функций. Со второго полугодия жизни мышечная гипотония заметно уменьшается, однако и у взрослых может сохраняться снижение мышечного тонуса. Появляется полифагия, развивается ожирение. Характерно отложение жира преимущественно в области туловища и проксимальных отделов конечностей, на этом фоне кисти и стопы кажутся диспропорционально маленькими (акромикрия). Акромикрия сочетается с клинодактилией, синдактилией, брахимезофалангией. Гипогонадотропный гипогонадизм у лиц мужского пола приобретает клиническую выраженность к пубертатному периоду и сохраняется у взрослых. Его особенности - резкое недоразвитие гениталий, скудное вторичное оволосение, снижение либидо и потенции, атрофия тестикулярной ткани, снижение сперматогенеза. Уже с рождения у мальчиков выявляют двусторонний крипторхизм, маленькую, гладкую мошонку и резкую гипоплазию полового члена, часто с фимозом. У лиц женского пола обнаруживают гипоплазию половых губ, позднее появление вторичных половых признаков, задержки менструаций вплоть до аменореи, инфантилизм матки, бесплодие. Больные обоих полов обычно стерильны. Психомоторное развитие детей замедлено, у большинства больных имеется различной формы умственная отсталость, в редких случаях отмечен нормальный или субнормальный интеллект. Больные, как правило, доброжелательны, безинициативны, плохо контролируют свои эмоции, им свойственна резкая смена настроения [6]. К специфическим черепно-лицевым дизморфиям относятся нерезко выраженная микроцефалия, гипоплазия хрящей ушных раковин, деформация и низкое расположение ушей, сдавленный в височных областях лоб, высокое арковидное небо, гипоплазия нижней челюсти, микродонтия с дефектами эмали и кариесом. Примерно у половины больных наблюдаются гипопигментация кожи, волос и радужки, некоторое повышение фоточувствительности. У 75% детей наблюдается гипопигментация кожи, волос и радужки. Часто диагностируется микроцефалия. Психомоторное развитие отстает от возрастной нормы - коэффициент интеллектуального развития - от 20 до 80 ед. (при норме 85-115 ед.). Речь затруднена, словарный запас уменьшен. Встречаются и другие аномалии: микродонтия, сколиоз, эктропион (выворот века), глаукома. При морфологическом исследовании мозга и ЯМР-томографии могут наблюдаться (примерно в 12% случаев) кисты червя мозжечка, аномалии коры головного мозга. Продолжительность жизни больных может достигать 60 лет и более. Согласно данным литературы, патогенез синдрома Прадера-Вилли до настоящего времени остается малоисследованным. Высказываются предположения, что ожирение у больных обусловлено значительным (более чем в 10 раз) усилением синтеза жира из ацетата и крайне низкими процессами липолиза. Гипогонадизм по гипогонадотропному типу может быть связан с дисфункцией гипоталамуса, преимущественно, в области вентромедиального и вентролатерального ядер. Правильность данной точки зрения подтверждается эффективностью лечения больных фармацевтическими препаратами (кломифен), приводившими к увеличению в плазме содержания лютеинизирующего гормона, тестостерона, нормализации показателей почечной экскреции гонадотропинов, сперматогенеза и появлению вторичных половых признаков [5]. Одним из объяснений гипопигментации кожи, волос и радужки служит снижение активности тирозиназы в волосяных фолликулах и меланоцитах, а также уменьшение пигмента в сетчатке. Обращается внимание на повышенный риск развития лейкемии у больных с синдромом Прадера-Вилли. Исследования выявили снижение репарации ДНК (до 65% по сравнению с 97% у здорового ребенка) в лимфоцитах больных с данной патологией. Не исключено, что низкая репарационная способность ДНК может оказать роковое влияние на развитие злокачественных новообразований у лиц с синдромом Прадера-Вилли [5]. Терапия синдрома Прадера-Вилли окончательно не разработана. По данным литературы, комплекс лечебных мероприятий включает лишь диету с ограничением жиров и углеводов и препараты, способствующие формированию вторичных половых признаков (гонадотропины). СПВ должен быть заподозрен у детей до 3-летнего возраста при наличии не менее 5 баллов, а у детей старше 3 лет - 8 баллов, при условии присутствия 4 и более больших признаков. Несмотря на высокую диагностическую чувствительность приведенной шкалы (около 90%), во всех случаях для подтверждения диагноза требуются кариотипирование и молекулярно-генетические исследования 15-й пары хромосом. Специфичность этих исследований достигает 100%. Кроме того, от выявленного типа генетических нарушений зависит генетический прогноз потомства. Дети, страдающие СПВ, должны постоянно находиться под наблюдением педиатра, невролога, психотерапевта, эндокринолога и офтальмолога.
Рис. 4. Девочка с синдромом Прадера-Вилли
|
||||
|
|
Последнее изменение этой страницы: 2020-03-26; просмотров: 88; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.190.176.176 (0.009 с.) |