
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Периодический Закон Д. И. МенделееваСодержание книги Поиск на нашем сайте
Закон Авогадро 1811 г. В равных объемах любых газов, взятых при одной и той же температуре и при одинаковом давлении, содержится одно и то же число молекул. Закон Эквивалентов Вещества взаимодействуют друг с другом в количествах, пропорциональных их эквивалентам. Другая формулировка (для решения задач) Массы(объемы)реагирующих друг с другом веществ пропорциональны их эквивалентным массам (объемам). Следствия из закона Гесса Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции (закон Лавуазье-Лапласа). Тепловой эффект химической реакции равен разности сумм теплот образования (ΔHf) продуктов реакции и исходных веществ, умноженных на стехиометрические коэффициенты (ν):
Тепловой эффект химической реакции равен разности сумм теплот сгорания (ΔHc) исходных веществ и продуктов реакции, умноженных на стехиометрические коэффициенты (ν):
Таким образом, пользуясь табличными значениями теплот образования или сгорания веществ, можно рассчитать теплоту реакции, не прибегая к эксперименту. Табличные величины теплот образования и сгорания веществ обычно относятся к т. н.стандартным условиям. Для расчёта теплоты процесса, протекающего при иных условиях, необходимо использовать и другие законы термохимии, например, закон Кирхгофа, описывающий зависимость теплового эффекта реакции от температуры. Если начальное и конечное состояния химической реакции (реакций) совпадают, то её (их) тепловой эффект равен нулю. Вопрос№5 Энтропия вещества из системы - мера беспорядка расположения в них частиц. Термодинамическая энтропия S, часто просто именуемая энтропия, в химии и термодинамике является функцией состояния термодинамической системы. Понятие энтропии было впервые введено в 1865 году Рудольфом Клаузиусом. Он определил изменение энтропии термодинамической системы при обратимом процессе как отношение общего количества тепла
Например, при температуре 0 °C, вода может находиться в жидком состоянии и при незначительном внешнем воздействии начинает быстро превращаться в лед, выделяя при этом некоторое количество теплоты. При этом температура вещества так и остается 0 °C. Изменяется состояние вещества, сопровождающееся выделением тепла, вследствие изменения структуры. Рудольф Клаузиус дал величине Эта формула применима только для изотермического процесса (происходящего при постоянной температуре). Её обобщение на случай произвольногоквазистатического процесса выглядит так:
где Необходимо обратить внимание на то, что рассматриваемое термодинамическое определение применимо только к квазистатическим процессам (состоящим из непрерывно следующих друг за другом состояний равновесия). Поскольку энтропия является функцией состояния, в левой части равенства стоит её полный дифференциал. Напротив, количество теплоты является функцией процесса, в котором эта теплота была передана, поэтому Энтропия, таким образом, согласно вышеописанному, определена вплоть до произвольной аддитивной постоянной. Третье начало термодинамики позволяет определить её точнее: предел величины энтропии равновесной системы при стремлении температуры к абсолютному нулю полагают равным нулю. Существует мнение, что мы можем смотреть на S и как на меру беспорядка в системе. В определённом смысле это может быть оправдано, потому что мы думаем об «упорядоченных» системах как о системах, имеющих очень малую возможность конфигурирования, а о «беспорядочных» системах как об имеющих очень много возможных состояний. Собственно, это просто переформулированное определение энтропии как числа микросостояний на данное макросостояние. Рассмотрим, например, распределение молекул идеального газа. В случае идеального газа наиболее вероятным состоянием, соответствующим максимуму энтропии, будет равномерное распределение молекул. При этом реализуется и максимальный «беспорядок», так как при этом будут максимальные возможности конфигурирования. Основные понятия: растворимость, растворитель, растворенное вещество. Растворимость – способность вещества растворяться в том или ином растворителе. Растворитель – компонент раствора, агрегатное состояние которого не изменяется при образовании раствора. Растворенное вещество – компоненты раствора за исключением растворителя. Коэффициент растворимости. Коэффициент растворимости – характеристика раствора, означающая число единиц массы безводного вещества, насыщающего при данных условиях 100 единиц массы растворителя. Измеряется в m (г) вещества на 100 г растворителя. ПДК. ПДК – максимальная концентрация вещества, не вызывающая токсического эффекта. Вопрос№15 Процесс растворения. Растворы не электролитов и электролитов. Электролиты и неэлектролиты. Растворы электролитов Вещества, распадающиеся на ионы в растворах или расплавах и потому проводящие электрический ток, называются электролитами. Так, хлорид натрия NaCl при растворении в воде полностью распадается на ионы натрия Na+ и хлорид-ионы Сl-. Вода образует ионы водорода Н+ и гидроксид-ионы ОН~ лишь в очень незначительных количествах. Вопрос№16 Свойства разбавленных растворов не электролитов. Законы Рауля и Вант-Гоффа о понижении давления над раствором, снижении температуры замерзания и повышений температуры кипения растворов, осмотическом давлении.
1. Понижение давления пара растворителя над раствором, Dр (закон Рауля) р1 = N1р0; Dр = р0 – р1 = N2po = Ро Здесь р1 - парциальное давление насыщенного пара растворителя над раствором; ро - давление насыщенного пара над чистым растворителем; N1 -мольная доля растворителя; N2 - мольная доля растворенного вещества; n1, -количество растворителя; n2 - количество растворенного вещества. 2. Понижение температуры кристаллизации раствора, Dtкрист Dtкрист = K × m. Здесь К - криоскопическая постоянная растворителя; m - моляльная концен-трация растворенного вещества. 3. Повышение температуры кипения раствора, Dtкип Dtкип = Е × m. Здесь Е- эбуллиоскопическая постоянная растворителя. 4. Осмотическое давление, Р, кПа P = CмRT. Здесь С м - молярная концентрация; R - газовая постоянная [8,31 Дж/моль×К)]; Т-температура, К. Первый закон Рауля Первый закон Рауля связывает давление насыщенного пара над раствором с его составом; он формулируется следующим образом: · Парциальное давление насыщенного пара компонента раствора прямо пропорционально его мольной доле в растворе, причём коэффициент пропорциональности равен давлению насыщенного пара над чистым компонентом.
Для бинарного раствора, состоящего из компонентов А и В (компонент А считаем растворителем) удобнее использовать другую формулировку: · Относительное понижение парциального давления пара растворителя над раствором не зависит от природы растворённого вещества и равно его мольной доле в растворе.
Растворы, для которых выполняется закон Рауля, называются идеальными. Идеальными при любых концентрациях являются растворы, компоненты которых очень близки по физическим и химическим свойствам (оптические изомеры, гомологи и т. п.), и образование которых не сопровождается изменением объёма и выделением либо поглощением теплоты. В этом случае силы межмолекулярного взаимодействия между однородными и разнородными частицами примерно одинаковы, и образование раствора обусловлено лишь энтропийным фактором. Правило Вант-Гоффа — эмпирическое правило, позволяющее в первом приближении оценить влияние температуры на скорость химической реакции в небольшом температурном интервале (обычно от 0 °C до 100 °C). Я. Х. Вант-Гофф на основании множества экспериментов сформулировал следующее правило:
Уравнение, которое описывает это правило, следующее:
где Следует помнить, что правило Вант-Гоффа применимо только для реакций с энергией активации 60-120 кДж/моль в температурном диапазоне 10-400oC. Правилу Вант-Гоффа также не подчиняются реакции, в которых принимают участие громоздкие молекулы, например, белки в биологических системах. Температурную зависимость скорости реакции более корректно описывает уравнение Аррениуса. Из уравнения Вант-Гоффа температурный коэффициент вычисляется по формуле:
Вопрос№17 Свойства разбавленных растворов электролитов. Степень диссоциации, константа диссоциации, изотопических коэффициент. Электролиты (от электро... и греч. lytos — разлагаемый, растворимый), жидкие или твёрдые вещества и системы, в которых присутствуют в сколько-нибудь заметной концентрации ионы, обусловливающие прохождение электрического тока. В узком смысле электролитами называются вещества, растворы которых проводят электрический ток ионами, образующимися в результате электролитической диссоциации. Электролиты в растворах подразделяют на сильные и слабые. Сильные электролиты практически полностью диссоциированы на ионы в разбавленных растворах. К ним относятся многие неорганические соли и некоторые неорганические кислоты и основания в водных растворах, а также в растворителях, обладающих высокой диссоциирующей способностью (спирты, амиды и др.). Молекулы слабых электролитов в растворах лишь частично диссоциированы на ионы, которые находятся в динамическом равновесии с недиссоциированными молекулами. К слабым электролитам относится большинство органических кислот и многие органические основания в водных и неводных растворах. Деление электролитов на сильные и слабые в некоторой степени условно, т. к. оно отражает не свойства самих электролитов, а их состояние в растворе. Последнее зависит от концентрации, природы растворителя, температуры, давления и др. По количеству ионов, на которые диссоциирует в растворе одна молекула, различают бинарные, или одно-одновалентные, электролиты (обозначаются 1-1электролиты, например КСl), одно-двухвалентные электролиты (обозначаются 1-2 электролиты, например CaCl2) и т. д. Электролиты типа 1-1, 2-2, 3-3 и т. п. называются симметричными, типа 1-2, 1-3 и т. п. — несимметричными. Свойства разбавленных растворов слабых электролитов удовлетворительно описываются классической теорией электролитической диссоциации. Для не слишком разбавленных растворов слабых электролитов, а также для растворов сильных электролитов эта теория неприменима, поскольку они являются сложными системами, состоящими из ионов, недиссоциированных молекул или ионных пар, а также более крупных агрегатов. Свойства таких растворовопределяются характером взаимодействий ион-ион, ион-растворитель, а также изменением свойств и структуры растворителя под влиянием растворённых частиц. Современные статистические теории сильных электролитов удовлетворительно описывают свойства лишь очень разбавленных (<0,1 моль/л)растворов. Электролиты чрезвычайно важны в науке и технике. Все жидкие системы в живых организмах содержат электролиты. Важный класс электролитов —полиэлектролиты. Электролиты являются средой для проведения многих химических синтезов и процессов электрохимических производств. При этом всё большую роль играют неводные растворы электролитов. Изучение свойств растворов электролитов важно для создания новых химических источников тока и совершенствования технологических процессов разделения веществ — экстракции из растворов и ионного обмена. Степень диссоциации — величина, характеризующая состояние равновесия в реакции диссоциации в гомогенных (однородных) системах. "Степень диссоциации это есть отношения числа продиссоциируемых молекул к общему числу молекул и умноженному на 100%":
числа распавшихся на ионы молекул к общему числу растворенных молекул. Степень диссоциации Константа диссоциации — вид константы равновесия, которая характеризует склонность объекта диссоциировать (разделяться) обратимым образом на частицы, как например когда комплексраспадается на составляющие молекулы, или когда соль диссоциирует в водном растворе на ионы. Константа диссоциации обычно обозначается Kd и обратна константе ассоциации. В случае с солями, константу диссоциации иногда называют константой ионизации. В общей реакции
где комплекс
где [A], [B] и [AxBy] — концентрации A, B и комплекса AxBy соответственно. Изотонический коэффициент (также фактор Вант-Гоффа; обозначается i) — безразмерный параметр, характеризующий поведение вещества в растворе. Он численно равен отношению значения некоторого коллигативного свойства раствора данного вещества и значения того же коллигативного свойства неэлектролита той же концентрации при неизменных прочих параметрах системы:
где solut. — данный раствор, nel. solut. — раствор неэлектролита той же концентрации, Tbp — температура кипения, а Tmp — температура плавления (замерзания). Вопрос№18 Законы Рауля и Вант-Гоффа для слабых электролитов. Законы Рауля
Найденные Вант-Гоффом значения i для растворов солей, кислот, оснований имеют разные значения, зависящие от природы и концентрации растворителя, и изменяются от 1 до 4.
Билет №19 Электролитическая диссоциация воды. Ионное произведение воды, водородный и другие показатели среды и методы их определения. Ио́нное произведе́ние воды́ — произведение концентраций ионов водорода Н+ и ионов гидроксида OH− в воде или в водных растворах, константа автопротолиза воды. Вывод значения ионного произведения воды Вода, хотя и является слабым электролитом, в небольшой степени диссоциирует: H2O + H2O ↔ H3O+ + OH−илиH2O ↔ H+ + OH− Равновесие этой реакции сильно смещено влево. Константу диссоциации воды можно вычислить по формуле:
Константа диссоциации воды при 25оС равна:
Эта величина постоянная при данной температуре (25оС), она называется ионным произведением воды KW:
Водородный показатель рН: В чистой воде при 25 °C концентрации ионов водорода ([H+]) и гидроксид-ионов ([OH−]) одинаковы и составляют 10−7 моль/л, это напрямую следует из определения ионного произведения воды, которое равно [H+] · [OH−] и составляет 10−14 моль²/л² (при 25 °C). Когда концентрации обоих видов ионов в растворе одинаковы, говорят, что раствор имеет нейтральную реакцию. При добавлении к воде кислоты концентрация ионов водорода увеличивается, а концентрация гидроксид-ионов соответственно уменьшается, при добавлении основания — наоборот, повышается содержание гидроксид-ионов, а концентрация ионов водорода падает. Когда [H+] > [OH−] говорят, что раствор является кислым, а при [OH−] > [H+] — щелочным. Для удобства представления, чтобы избавиться от отрицательного показателя степени, вместо концентраций ионов водорода пользуются их десятичным логарифмом, взятым с обратным знаком, который собственно и является водородным показателем — pH.
Билет №20 Обменные реакции в растворах электролитов. Условие необратимости реакций. В обменных реакциях, протекающих в растворах электролитов, наряду с недиссоциированными молекулами слабых электролитов, твердыми веществами и газами участвуют также находящиеся в растворе ионы. Поэтому сущность протекающих процессов наиболее полно выражается при записи их в форме ионно-молекулярных уравнений. Например, уравнения реакций нейтрализации сильных кислот щелочами HClO4 + NaOH →NaClO4 + H2O, 2HNO3 + Ba(OH)2 → Ba(NO3)2 + 2H2O, выражаются одним и тем же ионно-молекулярным уравнением H+ + OH– → H2O, из которого следует, что сущность этих процессов сводится к образованию из ионов водорода и гидроксид-ионов малодиссоциированного электролита – воды. Аналогично уравнения реакций BaCl2 +H2SO4 → BaSO4 + 2HCl, Ba(NO3)2 + Na2SO4 → BaSO4 + 2NaNO3 выражают один и тот же процесс образования из ионов Ва2+ и SO42— осадка малорастворимого электролита – сульфата бария Ва2+ + SO42– → BaSO4↓. На основании рассмотренных примеров можно сделать следующий вывод: реакции в растворах электролитов всегда идут в сторону образования наименее диссоциированных или наименее растворимых веществ. Из этого, в частности, следует, что сильные кислоты вытесняют слабые из растворов их солей Реакции в растворах электролитов идут до конца если в результате взаимодействия веществ происходит образование осадка, выделение газа и образование слабого электролита. При написании ионно - молекулярных уравнений реакций, слабые электролиты, малорастворимые соединения и газы записываются в молекулярной форме, а находящиеся в растворе сильные электролиты – в виде составляющих их ионов. Условия необратимости реакций (условия протекания реакций до конца): 1. Образование осадка. К2SО4 + BаСl2 ® BaSО4¯ + 2КСl 2К+ + SО42- + Bа2+ + 2Сl- ® BаSО4¯ + 2К+ + 2Сl- SО42- + Bа2- ® BаSО4¯ 2. Выделение газа. Na2S + 2HCl ® 2NaCl + H2S 2Na+ + S2- + 2H+ + 2Cl- ® 2Na+ + 2Cl- + H2S S2- + 2H+ ® H2S 3. Образование малодиссоциирующего соединения (слабого электролита или воды). NaOH + HCl ® NaCl + H2O Na+ + OH- + H+ + Cl- ® Na+ + Cl- + H2O OH- + H+ ® H2O 4. Образование комплексного соединения. NaOH + Al(OH)3 ® Na[Al(OH)4] Na+ + OH- + Al(OH)3 ® Na+ + [Al(OH)4]- OH- + Al(OH)3 ® [Al(OH)4]- Следовательно, реакции идут с образованием веществ с меньшей концентрацией ионов в растворе. В соответствии с законом действующих масс скорость реакции прямопропорциональна произведению концентрации реагирующих веществ. Следовательно, не возможность протекания обратной реакции в случае её необратимости объясняется тем, что концентрациях ионов в растворе уменьшается (ионы связываются в молекулы неэлектролитов), скорость обратной реакции стремится к нулю. Вывод: реакции в растворах электролитов могут протекать лишь только в том случае, если участвующие во взаимодействии ионы полностью или частично уходят из сферы реакции (в виде газа, осадка, слабого электролита или комплексного соединения). Билет №21 Гидролиз солями обратимый и не обратимый. Гидро́лиз (от др.-греч. ὕδωρ — вода + λύσις — разложение) — один из видов химических реакций сольволиза, где при взаимодействии веществ с водой происходит разложение исходного вещества с образованием новых соединений. Механизм гидролиза соединений различных классов: соли, углеводы, белки, сложные эфиры, жиры и др. имеет существенные различия. Гидролиз солей — разновидность реакций гидролиза, обусловленного протеканием реакций ионного обмена в растворах (преимущественно, водных) растворимых солей-электролитов. Движущей силой процесса является взаимодействие ионов с водой, приводящее к образованию слабого электролита в ионном или (реже) молекулярном виде («связывание ионов»). Различают обратимый и необратимый гидролиз солей[1]: · 1. Гидролиз соли слабой кислоты и сильного основания (гидролиз по аниону):
(раствор имеет слабощелочную среду, реакция протекает обратимо, гидролиз по второй ступени протекает в ничтожной степени) · 2. Гидролиз соли сильной кислоты и слабого основания (гидролиз по катиону):
(раствор имеет слабокислую среду, реакция протекает обратимо, гидролиз по второй ступени протекает в ничтожной степени) · 3. Гидролиз соли слабой кислоты и слабого основания:
(равновесие смещено в сторону продуктов, гидролиз протекает практически полностью, так как оба продукта реакции уходят из зоны реакции в виде осадка или газа). ·
Билет №22 Электрохимические системы. Окислительно-восстановительные реакции. Составление уравнений. Электронный баланс. Существование электрохимических систем возможно из-за возникновения разности потенциалов между металлами и электролитом при их контакте. Измерить потенциал металла (электрода) непосредственно нельзя, но можно измерить его относительно другого электрода. Эталоном при сопоставлении металлов по их энергетическому потенциалу является стандартный водородный электрод, потенциал которого условно принимается за нуль. Его устройство таково: платиновый электрод покрыт мелкодисперсной платиной (платиновой чернью), погружен в раствор серной кислоты с концентрацией ионов водорода 1 моль/л, обдувается струей газообразного водорода под давлением 100 кПа (Т = 298 K). Водород адсорбируется на поверхности платины. На практике при потенциометрических измерениях водородный электрод используют редко. Чаще применяют более удобные компактные электроды сравнения, имеющие определенное значение потенциала относительно водородного электрода. Обычно пользуются каломельным электродом, состоящим из металлической ртути и раствора хлорида ртути (каломели Hg2Cl2) в хлориде калия. Потенциал каломельного электрода зависит от концентрации ионов ртути, а последняя – от концентрации раствора KCl. На основании теоретических расчетов установлено, что величина электродного потенциала, возникающая на границе между металлом и раствором соли этого металла (т. е. раствором, содержащим ионы этого металла), равна:
где Е 0 – электрическая постоянная, зависящая от выбора электрода сравнения, R – газовая постоянная, равная 8,32 Дж/граджмоль, Т – абсолютная температура, n – степень окисления металла в данном соединении (в соответствии с теорией строения атома – число электронов, которое теряет атом металла, превращаясь в ион), F – число Фарадея, с – молярная концентрация ионов металла в данном растворе. Окисли́тельно-восстанови́тельные реа́кции, также редокс (англ. redox, от red uction- ox idation — окисление-восстановление) — это встречно-параллельные химические реакции, протекающие с изменением степеней окисления атомов, входящих в состав реагирующих веществ, реализующихся путём перераспределения электронов между атомом-окислителем и атомом-восстановителем. Окислительно-восстановительная реакция между водородом и фтором
Разделяется на две полуреакции: 1) Окисление:
2) Восстановление:
Окисление, восстановление В окислительно-восстановительных реакциях электроны от одних атомов, молекул или ионов переходят к другим. Процесс отдачи электронов — окисление. При окислении степень окисления повышается:
Процесс присоединения электронов — восстановление. При восстановлении степень окисления понижается:
Метод электронного баланса складывается из следующих этапов: а) записывают схему реакции (формулы реагентов и продуктов), а затем находят элементы, которые повышают и понижают свои степени окисления, и выписывают их отдельно: MnCO3 + KClO3 → MnO2 + KCl + CO2 ClV → Cl−I MnII → MnIV б) составляют уравнения полуреакций восстановления и окисления, соблюдая законы сохранения числа атомов и заряда в каждой полуреакции: полуреакция восстановления ClV + 6e− = Cl−I полуреакция окисления MnII − 2e− = MnIV в) подбирают дополнительные множители для уравнения полуреакций так, чтобы закон сохранения заряда выполнялся для реакции в целом, для чего число принятых электронов в полуреакциях восстановления делают равным числу отданных электронов в полуреакции окисления: ClV + 6e− = Cl−I * 1 MnII − 2e− = MnIV * 3 г) проставляют (по найденным множителям) стехиометрические коэффициенты в схему реакции (коэффициент 1 опускается): 3MnCO3 + KClO3 = 3MnO2 + KCl + CO2 д) уравнивают числа атомов тех элементов, которые не изменяют своей степени окисления при протекании реакции (если таких элементов два, то достаточно уравнять число атомов одного из них, а по второму провести проверку). Получают уравнение химической реакции: 3MnCO3 + KClO3 = 3MnO2 + KCl + 3CO2 е) проводят проверку по элементу, который не менял свою степень окисления (чаще всего это кислород): слева 9 + 3 = 12 атомов O справа 6 + 6 = 12 атомов O Подбор коэффициентов проведен правильно. Билет №23 Механизм образования двойного электрического слоя и электродного потенциала на границе раздела металл-электролит.
Билет №24 Зависимость электродного потенциала от природы электродов, температуры, концепции потенциала-определяющих ионов. Уравнение Нериста.
Билет №25 Типы электродов (металлические, газовые, первого и второго родов). К электродам первого рода относятся такие, потенциал которых относительно какого-либо электрода сравнения определяется концентрацией катионов. К ним принадлежат металлы, погруженные в растворы своих солей. Разновидностью электродов первого рода являются амальгамные электроды, т.е. состоящие либо из растворов металлов в ртути, либо вообще из сплавов металлов жидких или твердых растворов. В этом случае процесс на электроде изображается уравнением Men + + ne и, следовательно:
Отсюда следует, что потенциал амальгамного электрода зависит от активности катиона в растворе и активности компонента в металлической фазе. Электроды второго рода обычно имеют следующее устройство. Металл погружен в насыщенный раствор своей малорастворимой соли, в котором находится другая хорошо растворимая соль с тем же анионом. Примером может служить электрод из металлического серебра, находящегося в соприкосновении с осадком AgCl, т.е. в растворе, насыщенном этой солью. Этот раствор также должен содержать и другой хорошо растворимый электролит с одноименным ионом (например, KCl). На электроде происходит обратимая реакция перехода ионов серебра в раствор или их разрядки, т.е. Agт ←→ Ag+ + e. За этим следует реакция Ag+ + Cl- = AgClт. Суммарный процесс описывается уравнением Agт + Cl- = AgClт + e. Так как a AgClт = 1, то, согласно уравнению (IX.20), E = E 0 - | |||||||||||||||||||
|
| Поделиться: |

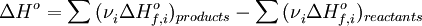

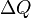 к величине абсолютной температуры
к величине абсолютной температуры  :
: .
. имя «энтропия», происходящее от греческого слова τρoπή, «изменение» (изменение, превращение, преобразование). Данное равенство относится к изменению энтропии, не определяя полностью саму энтропию.
имя «энтропия», происходящее от греческого слова τρoπή, «изменение» (изменение, превращение, преобразование). Данное равенство относится к изменению энтропии, не определяя полностью саму энтропию. ,
, — приращение (дифференциал) энтропии некоторой системы, а
— приращение (дифференциал) энтропии некоторой системы, а  — бесконечно малое количество теплоты, полученное этой системой.
— бесконечно малое количество теплоты, полученное этой системой.


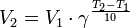
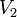 — скорость реакции при температуре
— скорость реакции при температуре  ,
, 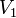 — скорость реакции при температуре
— скорость реакции при температуре  ,
,  — температурный коэффициент реакции (если он равен 2, например, то скорость реакции будет увеличиваться в 2 раза при повышении температуры на 10 градусов).
— температурный коэффициент реакции (если он равен 2, например, то скорость реакции будет увеличиваться в 2 раза при повышении температуры на 10 градусов).

 равна отношению числа диссоциированных молекул
равна отношению числа диссоциированных молекул  к сумме
к сумме  , где
, где  — число недиссоциированных молекул. Часто
— число недиссоциированных молекул. Часто 
 разбивается на x единиц A и y единиц B, константа диссоциации определяется так:
разбивается на x единиц A и y единиц B, константа диссоциации определяется так:
 ,
, Закон Вант-Гоффа
Закон Вант-Гоффа



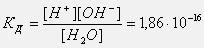 Такое значение константы соответствует диссоциации одной из ста миллионов молекул воды, поэтому концентрацию воды можно считать постоянной и равной 55,55 моль/л (плотность воды 1000 г/л, масса 1 л 1000 г, количество вещества воды 1000г:18г/моль=55,55 моль, С=55,55 моль: 1 л = 55,55 моль/л). Тогда
Такое значение константы соответствует диссоциации одной из ста миллионов молекул воды, поэтому концентрацию воды можно считать постоянной и равной 55,55 моль/л (плотность воды 1000 г/л, масса 1 л 1000 г, количество вещества воды 1000г:18г/моль=55,55 моль, С=55,55 моль: 1 л = 55,55 моль/л). Тогда








 4. Соль сильной кислоты и сильного основания не подвергается гидролизу, и раствор нейтрален.
4. Соль сильной кислоты и сильного основания не подвергается гидролизу, и раствор нейтрален.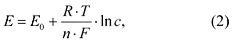



















 уравнение Нернста
уравнение Нернста




