
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Белки,строение,биологическая рольСодержание книги
Поиск на нашем сайте
Белки,строение,биологическая роль Белки — высокомолекулярные органические соединения, состоящие из остатков α-аминокислот. В состав белков входят углерод, водород, азот, кислород, сера. Часть белков образует комплексы с другими молекулами, содержащими фосфор, железо, цинк и медь. Бесконечное разнообразие белков создается за счет различного сочетания всего 20 аминокислот. Каждая аминокислота имеет свое название, особое строение и свойства. Их общую формулу можно представить в следующем виде:
Молекула аминокислоты состоит из двух одинаковых для всех аминокислот частей, одна из которых является аминогруппой (—NH2) с основными свойствами, другая — карбоксильной группой (—COOH) с кислотными свойствами. Часть молекулы, называемая радикалом (R), у разных аминокислот имеет различное строение. Наличие в одной молекуле аминокислоты основной и кислотной групп обусловливает их высокую реакционную способность. через эти группы происходит соединение аминокислот при образовании белка. При этом возникает молекула воды, а освободившиеся электроны образуют пептидную связь. Поэтому белки называют полипептидами.
Биологическая роль белков: 1) защитная (интерферон усиленно синтезируется в организме при вирусной инфекции); 2) структурная (коллаген входит в состав тканей, участвует в образовании рубца); 3) двигательная (миозин участвует в сокращении мышц); 4) запасная (альбумины яйца); 5) транспортная (гемоглобин эритроцитов переносит питательные вещества и продукты обмена); 6) рецепторная (белки-рецепторы обеспечивают узнавание клеткой веществ и других клеток); 7) регуляторная (регуляторные белки определяют активность генов); 8) белки-гормоны участвуют в гуморальной регуляции (инсулин регулирует уровень сахара в крови); 9) белки-ферменты катализируют все химические реакции в организме; 10) энергетическая (при распаде 1 г белка выделяется 17 кдж энергии).
Гликоген. Синтез гликогена Гликоген — депонированная форма глюкозы, высвобождает эту гекеозу при участии гликогенфосфорилазы. Фермент катализирует фосфоролиз (расщепление с присоединением компонентов фосфорной кислоты) 1,4-гликозидной связи, с высвобождением остатков глюкозы в виде глюкозо-1-фосфата (Г-1-Ф), который под действием фосфоглюкомутазы превращается в Г-6-Ф. Его возможные пути превращения"
1) в мышцах, где нет глюкозо-6-фосфатазы, по основному пути (аэробному или анаэробному); 2) в жировой ткани и других, где идут интенсивные восстановительные синтезы, по пентозофосфатному пути (для накопления НАДФ • Нд); 3) в печени, где много глюкозо-6-фосфатазы, расщепляется на глюкозу и фосфат, глюкоза поступает в кровь. Синтез: 1. глюкоза подвергается фосфорилированию при участии фермента гексокиназы, а в печени – и глюкокиназы. Далее глюкозо-6-фосфат под влиянием фермента фосфоглюкомутазы переходит в глюкозо-1-фос-фат:
Образовавшийся глюкозо-1-фосфат уже непосредственно вовлекается в синтез гликогена. На первой стадии синтеза глюкозо-1-фосфат вступает во взаимодействие с УТФ (уридинтрифосфат), образуя уридиндифосфатглю-козу (УДФ-глюкоза) и пирофосфат. Данная реакция катализируется ферментом глюкозо-1-фосфат-уридилилтрансферазой (УДФГ-пирофосфорила-за): Глюкозо-1-фосфат + УТФ < = > УДФ-глюкоза + Пирофосфат.
2. На второй стадии – стадии образования гликогена – происходит перенос глюкозного остатка, входящего в состав УДФ-глюкозы, на глюкозидную цепь гликогена («затравочное» количество). При этом образуется α-(1–>4)-связь между первым атомом углерода добавляемого остатка глюкозы и 4-гидроксильной группой остатка глюкозы цепи. Эта реакция катализируется ферментом гликогенсинтазой. Необходимо еще раз подчеркнуть, что реакция, катализируемая гликогенсинтазой, возможна только при условии, что полисахаридная цепь уже содержит более 4 остатков D-глюкозы.
Образующийся УДФ затем вновь фосфорилируется в УТФ за счет АТФ, и таким образом весь цикл превращений глюкозо-1-фосфата начинается сначала.
Тканевое дыхание Это процесс потребление клетками тканей организма кислорода, который участвует в биологическом окислении. Такой вид окисления называют аэробным окислением. Если конечным акцептором в цепи переноса водорода выступает не кислород, а другие вещества (например пировиноградная кислота), то такой тип окисления называют анаэробным. Т.о. биологическое окисление - это дегидрирование субстрата с помощью промежуточных переносчиков водорода и его конечного акцептора.
Особенности тканевого дыхания Процесс тканевого дыхания нельзя считать тождественным процессам биологического окисления (ферментативным процессам окисления различных субстратов, протекающим в животных, растительных и микробных клетках), поскольку значительная часть таких окислительных превращений в организме происходит в анаэробных условиях, т.е. без участия молекулярного кислорода, в отличие от дыхания тканей. Большая часть энергии в аэробных клетках образуется благодаря дыханию тканей, и количество образующейся энергии зависит от его интенсивности. Интенсивность Д. т. определяется скоростью поглощения кислорода на единицу массы ткани; в норме она обусловлена потребностью ткани в энергии. Интенсивность его наиболее высока в сетчатке глаза, почках, печени; она значительна в слизистой оболочке кишечника, щитовидной железе, яичках, коре головного мозга, гипофизе, селезенке, костном мозге, легких, плаценте, вилочковой железе, поджелудочной железе, диафрагме, сердце, скелетной мышце, находящейся в состоянии покоя. В коже, роговице и хрусталике глаза интенсивность тканевого дыхания невелика. Гормоны щитовидной железы, жирные кислоты и другие биологически активные вещества способны активизировать тканевое дыхание.
Оксилипиды - Оксилипиды липоксигеназного пути - Оксилипиды циклооксигеназного пути
24. Холестери́н.Биологическое значение для организма.Атеросклероз Холестери́н (др.-греч. χολή — желчь и στερεός — твёрдый; синоним: холестерол) — органическое соединение, природный жирный (липофильный) спирт, содержащийся в клеточных мембранах всех живых организмов за исключением безъядерных (прокариоты). Нерастворим в воде, растворим в жирах и органических растворителях. Около 80 % холестерина вырабатывается самим организмом (печенью,кишечником, почками, надпочечниками, половыми железами), остальные 20 % поступают с пищей. В организме находится 80 % свободного и 20 % связанного холестерина. Холестерин обеспечивает стабильность клеточных мембран в широком интервале температур. Он необходим для выработки витамина D, выработки надпочечниками различных стероидных гормонов, включая кортизол, альдостерон, женских половых гормонов эстрогенов и прогестерона, мужского полового гормона тестостерона, а по последним данным — играет важную роль в деятельности синапсов головного мозга и иммунной системы, включая защиту от рака Биологическая роль: Холестерин в составе клеточной плазматической мембраны играет роль модификатора бислоя, придавая ему определённую жёсткость за счёт увеличения плотности «упаковки» молекул фосфолипидов. Таким образом, холестерин — стабилизатор текучести плазматической мембраны Холестерин открывает цепь биосинтеза стероидных половых гормонов и кортикостероидов, служит основой для образования жёлчных кислот и витаминов группы D, участвует в регулировании проницаемости клеток и предохраняет эритроциты крови от действия гемолитических ядов. Холестерин нерастворим в воде и в чистом виде не может доставляться к тканям организма при помощи основанной на воде крови. Вместо этого холестерин в крови находится в виде хорошо растворимых комплексных соединений с особыми белками-транспортерами, так называемыми аполипопротеинами. Такие комплексные соединения называются липопротеинами.
Существует несколько видов аполипопротеинов, различающихся молекулярной массой, степенью сродства к холестерину и степенью растворимости комплексного соединения с холестерином (склонностью к выпадению кристаллов холестерина в осадок и к формированию атеросклеротических бляшек). Различают следующие группы: высокомолекулярные (HDL, ЛПВП, липопротеины высокой плотности) и низкомолекулярные (LDL, ЛПНП, липопротеины низкой плотности), а также очень низкомолекулярные (VLDL, ЛПОНП, липопротеины очень низкой плотности) и хиломикрон. К периферийным тканям холестерин транспортируется хиломикроном, ЛПОНП и ЛПНП. К печени, откуда затем холестерин удаляется из организма, его транспортируют аполипротеины группы ЛПВП. Нарушения липидного обмена считаются одним из наиболее важных факторов развития атеросклероза. К атерогенным нарушениям липидного обмена относятся: 1Повышение уровня общего холестерина крови 2Повышение уровня триглицеридов и липопротеинов низкой плотности (ЛНП) 3Снижение уровня липопротеинов высокой плотности (ЛВП). Связь повышенного уровня холестерина и атеросклероза неоднозначна: с одной стороны увеличение содержания холестерина в плазме крови считается бесспорным фактором риска атеросклероза, с другой стороны атеросклероз часто развивается у людей с нормальным уровнем холестерина. В действительности высокий уровень холестерина является лишь одним из многочисленных факторов риска атеросклероза (ожирение,курение, диабет, гипертония). Наличие этих факторов у людей с нормальным уровнем холестерина потенцирует негативное влияние свободного холестерина на стенки сосудов и тем самым приводит к образованию атеросклероза при более низких концентрациях холестерина в крови. Существует также иной взгляд на проблематику холестерина. Холестерин как «ремонтный» материал скапливается в местах микроповреждений сосудов и блокирует эти повреждения, выполняя гомогенную лекарственную роль. Именно поэтому атеросклероз наблюдается у людей с нормальным уровнем холестерина. У людей с повышенным уровнем проблема появляется быстрее, плюс, наличие повышенного уровня холестерина проще статистически связать с атеросклерозом, что и было сделано в начале исследований, из-за чего холестерин был объявлен виновником всех бед. Поэтому же, просто снижение уровня холестерина само по себе не решает всех проблем с сосудами. Недостаток холестерина в таком случае может явиться причиной кровоизлияний. Требуется дальнейшее изучение причин, вызывающих повреждения сосудов и разработка методов их лечения.
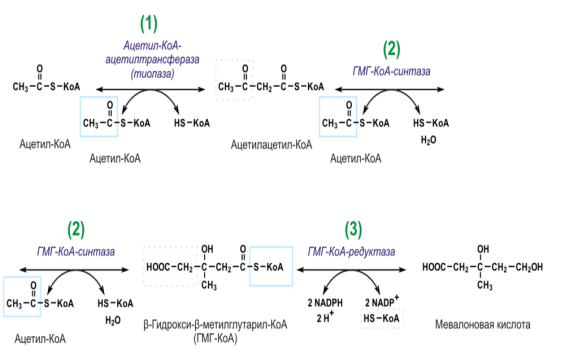
25. Cинтез холестерина до мевалоновой кислоты Cинтез мевалоната протекает в три этапа. 1. Образование ацетоацетил-КоА из двух молекул ацетил-КоА с помощью тиолазного фермента ацетоацетилтрансферазы. Реакция обратима. Происходит в цитозоле. 2. Образование β-гидрокси-β-метилглутарил-КоА из ацетоацетил-коА с третьей молекулой ацетил-КоА с помощью гидроксиметилглутарил-КоА-синтазы (ГМГ-КоА-синтазы). Реакция также обратима. Происходит в цитозоле. 3. Образование мевалоната восстановлением ГМГ и отщеплением HS-KoA с помощью НАДФ-зависимой гидроксиметилглутарил-КоА-редуктазы (ГМГ-КоА-редуктаза). Происходит в гЭПР. Это первая практически необратимая реакция в цепи биосинтеза холестерина, а также она лимитирует скорость биосинтеза холестерина. Отмечены суточные колебания синтеза этого фермента. Активность его увеличивается при введении инсулина и тиреоидных гормонов, снижается при голодании, введении глюкагона, глюкокортикоидов.
Обмен липидов в ЖКТ В процессах пищеварения все омыляемые липиды (жиры, фосфолипиды, гликолипиды, стериды) подвергаются гидролизу на составные части. В составе липидов пищи преобладают триглицериды. Большая часть поступающих с пищей триглицеридов расщепляется до моноглицеридов и жирных кислот в тонком кишечнике. Гидролиз жиров происходит под влиянием липаз сока поджелудочной железы и слизистой оболочки тонкого кишечника. Соли желчных кислот и фосфолипиды, проникающие из печени в просвет тонкого кишечника в составе желчи, способствуют образованию устойчивых эмульсий. В результате эмульгирования резко увеличивается площадь соприкосновения образовавшихся мельчайших капелек жира с водным раствором липазы, и этим самым увеличивается липолитическое действие фермента. Соли желчных кислот стимулируют процесс расщепления жиров не только участвуя в их эмульгировании, но и активируя липазу. Расщепление стероидов происходит в кишечнике при участии фермента холинэстеразы, выделяющегося с соком поджелудочной железы. В результате гидролиза стероидов образуются жирные кислоты и холестерин. Фосфолипиды расщепляются полностью или частично под действием гидролитических ферментов - специфических фосфолипаз. Продуктом полного гидролиза фосфолипидов являются: глицерин, высшие жирные кислоты, фосфорная кислота и азотистые основания. Всасыванию продуктов переваривания жиров предшествует образование мицелл - надмолекулярных образований или ассоциатов. Мицеллы содержат в качестве основного компонента соли желчных кислот, в которых растворены жирные кислоты, моноглицериды, холестерин и т.п. В клетках кишечной стенки из продуктов пищеварения, а в клетках печени, жировой ткани и других органов из предшественников, возникших в обмене углеводов и белков, происходит построение молекул специфических липидов тела человека - ресинтез триглицеридов и фосфолипидов. Однако их жирнокислотный состав по сравнению с жирами пищи изменен: в триглицеридах, синтезируемых в слизистой оболочке кишечника содержатся арахидоновая и линоленовая кислоты даже в том случае, если они отсутствуют в пище. Кроме того, в клетках кишечного эпителия жировая капля покрывается белковой оболочкой и происходит формирование хиломикронов - большая жировая капля, окруженная небольшим количеством белка. Транспортирует экзогенные липиды в печень, адипозную ткань, соединительную ткань, в миокард. Поскольку липиды и некоторые их составные части нерастворимы в воде, для переноса из одного органа в другой они образуют особые транспортные частицы, в составе которых обязательно есть белковый компонент. В зависимости от места образования эти частицы различаются структурой, соотношением составных частей и плотностью. Если в составе такой частицы в процентном соотношении жиры преобладают над белками, то такие частицы называются липопротеинами очень низкой плотности (ЛПОНП) или липопротеинами низкой плотности (ЛПНП). По мере увеличения процентного содержания белка (до 40%) частица превращается в липопротеин высокой плотности (ЛПВП). В настоящее время изучение таких транспортных частиц дает возможность с большой степенью точности оценивать состояние липидного обмена организма и использование липидов в качестве источников энергии.
Если образование липидов происходит из углеводов или белков, предшественником глицерина становится промежуточный продукт гликолиза - фосфодиоксиацетон, жирных кислот и холестерина - ацетилкофермент А, аминоспиртов - некоторые аминокислоты. Синтез липидов требует больших энерготрат для активации исходных веществ. Основной часть продуктов распада жиров всасывается из клеток кишечного эпителия в лимфатическую систему кишечника, грудной лимфатический проток и только затем - в кровь.
Патологии ЛИПИДНОГО ОБМЕНА Нарушение процессов всасывания жиров. Нарушения липидного обмена возможны уже в процессе переваривания и всасывания жиров. Одна группа расстройств связана с недостаточным поступлением панкреатической липазы в кишечник, вторая обусловлена нарушением поступления в кишечник желчи. Кроме того, нарушения процессов переваривания и всасывания липидовмогут быть связаны с заболеваниями пищеварительного тракта (при энтеритах, гиповитаминозах и некоторых других патологических состояниях). Образовавшиеся в полости кишечника моноглицериды и жирные кислоты не могут нормально всасываться вследствие повреждения эпителиального покрова кишечника. Во всех этих случаях кал содержит много нерасщепленного жира или невсосавшихся высших жирных кислот и имеет характерный серовато-белый цвет. Нарушение процессов перехода жира из крови в ткань. При недостаточной активности липопротеинлипазы крови нарушается переход жирных кислот из хиломикронов (ХМ) плазмы крови в жировые депо (не расщепляются триглицериды). Чаще это наследственное заболевание, обусловленное полным отсутствием активности липопротеинлипазы. Плазма крови при этом имеет молочный цвет в результате чрезвычайно высокого содержания ХМ. Наиболее эффективным лечением этого заболевания является замена природных жиров, содержащих жирные кислоты с 16–18 углеродными атомами, синтетическими, в состав которых входят короткоцепочечные жирные кислоты с 8–10 углеродными атомами. Эти жирные кислоты способны всасываться из кишечника непосредственно в кровь без предварительного образования ХМ. Кетонемия и кетонурия. В крови здорового человека кетоновые (ацетоновые) тела содержатся в очень небольших концентрациях. Однако при голодании, а также у лиц с тяжелой формой сахарного диабета содержание кетоновых тел в крови может повышаться до 20 ммоль/л. Это состояние носит название кетонемии; оно обычно сопровождается резким увеличением содержания кетоновых тел в моче (кетонурия). Например, если в норме за сутки с мочой выводится около 40 мг кетоновых тел, то при сахарном диабете содержание их в суточной порции мочи может доходить до 50 г и более. В настоящее время явления кетонемии и кетонурии при сахарном диабете или голодании можно объяснить следующим образом. И диабет, и голодание сопровождаются резким сокращением запасов гликогена в печени. Многие ткани и органы, в частности мышечная ткань, находятся в состоянии энергетического голода (при недостатке инсулина глюкоза не может с достаточной скоростью поступать в клетку). В этой ситуации благодаря возбуждению метаболических центров в ЦНС импульсами с хе-морецепторов клеток, испытывающих энергетический голод, резко усиливаются липолиз и мобилизация большого количества жирных кислот из жировых депо в печень. В печени происходит интенсивное образование кетоновых тел. Образующиеся в необычно большом количестве кетоновые тела (ацетоуксусная и β-гидроксимасляная кислоты) с током крови транспортируются из печени к периферическим тканям. Периферические ткани при диабете и голодании сохраняют способность использовать кетоновые тела в качестве энергетического материала, однако ввиду необычно высокой концентрации кетоновых тел в притекающей крови мышцы и другие органы не справляются с их окислением и как следствие возникает кетонемия. Атеросклероз и липопротеины. В настоящее время доказана ведущая роль определенных классов липопротеинов в патогенезе атеросклероза. Известное положение акад. Н.Н. Аничкова «без холестерина нет атеросклероза» с учетом современных знаний можно выразить иначе: «без атерогенных липопротеинов не может быть атеросклероза».
27. Жирные кислоты.Бета-окисление жирных кислот Жирные кислоты — алифатические одноосновные карбоновые кислоты с открытой цепью, содержащиеся в этерифицированной форме в жирах, маслах и восках растительного и животного происхождения. Жирные кислоты, как правило, содержат неразветвленную цепь из четного числа атомов углерода (С4-24, включая карбоксильный углерод) и могут быть как насыщенными, так и ненасыщенными. Ненасыщенные жирные кислоты в свою очередь делятся на а) моноеновые те содержащие одну двойную связь б) полиеновые, содержащие много двойных связей (диеновые, триеновые и др) Природные ненасыщенные жирные кислоты (незаменимые) обычно имеют тривиальное название, например алеиновая, линоливая, линоленовая арахндоновая Жирные кислоты в организме выполняют несколько функций. Прежде всею несомненно это энергетическая функция. Так же выполняют структурную функцию. Выполняют пластическую функцию. Процесс β-окисления протекает поэтапно. На каждом этапе от жирной кислоты отщепляется двухуглеродный фрагмент в виде ацетил-коэнзима А, а также происходит восстановление НАД+ до НАД∙Н и ФАД до ФАД∙Н2. В ходе первой реакции происходит окисление группы –СН2-СН2–, расположенной около карбонильного атома углерода. Как и при окислении сукцината в цикле Кребса, окислителем служит ФАД. Затем (вторая реакция) происходит гидратация двойной связи образовавшегося непредельного соединения, при этом третий атом углерода становится гидроксилированным – образуется β-оксикислота, присоединенная к коэнзиму А. В ходе третьей реакции происходит окисление этой спиртовой группы до кетогруппы, в качестве окислителя используется НАД+. Наконец, с образовавшимся β-кетоацил-коэнзимом А реагирует другая молекула коэнзима А. В результате отщепляется ацетил-коэнзим А, и ацил-КоА укорачивается на два углеродных атома. Теперь циклический процесс будет протекать по второму заходу, остаток жирной кислоты укоротится еще на один ацетил-КоА, и так до полного расщепления жирной кислоты. Из четырех реакций β-окисления только первая является необратимой, остальные – обратимы, их прохождение слева направо обеспечивается постоянным выводом конечных продуктов. Суммарно β-окисление пальмитоил-коэнзима А протекает согласно уравнению: C15H31CO-КоА + 7НАД+ + 7ФАД + 7КоА + 7Н2О = 8ацетил-КоА + 7НАД∙Н + 7ФАД∙Н2 + 7Н+ Ацетил-КоА затем поступает в цикл Кребса. НАД∙Н и ФАД∙Н2 окисляются в митохондриях, обеспечивая энергией синтез АТФ.
29. Желчные кислоты, строение, биологическая роль. Желчные кислоты - тетрациклические монокарбоновые оксикислоты из класса стероидов. По химической природе являются производными холановой кислоты С23Н39СООН. Они -конечный продукт метаболизма холестерина. Желчные кислоты образуются в печени и выделяются с желчью, как в свободном виде, так и как парные соединения с глицином и таурином. Глицин и таурин связаны с желчными кислотами пептидными связями. В желчи человека в основном содержатся холевая, дезоксихолевая и хенодезоксихолевая. Кроме того, в малых количествах присутствуют литохолевая, аллохолевая и уреодезоксихолевые кислоты. После выделения желчи в кишечник при действии ферментов кишечной микрофлоры из первичных желчных кислот образуются литохолевая и дезоксихолевая кислоты - вторичные желчные кислоты. Они всасываются из кишечника, с кровью воротной вены попадают в печень, а затем в желчь. Желчные кислоты обладают амфифильными свойствами. Боковая цепь с остатком глицина или таурина гидрофильна, а циклическая часть является гидрофобной. Амфифильная природа желчных кислот обусловливает их участие в переваривании и всасывании жиров. Желчные кислоты являются поверхностно-активными веществами, принимают участие в эмульгировании жиров. Желчные кислоты резко уменьшают поверхностное натяжение на границе жир/вода. Эмульгирование жиров ускоряет процессы переваривания липидов, т.к. увеличивается поверхность соприкосновения жира с липазой поджелудочной железы. Наиболее мощное эмульгирующее действие на жиры оказывают щелочные (натриевые или калиевые) соли парных желчных кислот. Желчные кислоты являются активаторами липолитических ферментов (превращение пролипазы в липазу), повышают активность панкреатической липазы в 10-15 раз; а также регулируют перистальтику (моторику) кишечника, обладают бактерицидным действием, подавляя гнилостные процессы. Желчные кислоты принимают участие во всасывании жиров. Они образуют с жирными кислотами и холеиновые комплексы, которые проникают в клетки слизистой кишечника. Отсюда желчные кислоты поступают в кровь, а с ней - в печень, повторно участвуя в образовании желчи (90-95 % проходят энтерогепатический цикл 5-10 раз за сутки). Небольшая часть желчных кислот - около 0,5 г за сутки - выводится из организма. Фонд желчных кислот обновляется полностью примерно за 10 дней. ДЕЗАМИНИРОВАНИЕ. БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ. ПРИМЕРЫ. Доказано существование 4 типов дезаминирования аминокислот (отщепление аминогруппы). Выделены соответствующие ферментные системы, катализирующие эти реакции, и идентифицированы продукты реакции. Во всех случаях NH2-группа аминокислоты освобождается в виде аммиака.
Помимо аммиака, продуктами дезаминирования являются жирные кислоты, оксикислоты и кетокислоты. Для животных тканей, растений и большинства аэробных микроорганизмов преобладающим типом реакций является окислительное дезаминирование аминокислот, за исключением гис-тидина, подвергающегося внутримолекулярному дезаминированию. Рассмотрим более подробно механизм окислительного дезаминиро-вания аминокислот, протекающего в две стадии.
Первая стадия является ферментативной и завершается образованием неустойчивого промежуточного продукта (иминокислота), который на второй стадии спонтанно без участия фермента, но в присутствии воды распадается на аммиак и α-кетокислоту. Следует указать, что оксидазы аминокислот (L- и D-изомеров) являются сложными флавопротеинами, содержащими в качестве кофермента ФМН или ФАД, которые выполняют в этой реакции роль акцепторов двух электронов и протонов, отщепляющихся от аминокислоты. Оксидазы L-аминокислот могут содержать как ФМН, так и ФАД, а оксидазы D-аминокислот – только ФАД в качестве простетической группы. Схематически реакции окислительного дезами-нирования аминокислот с участием коферментов могут быть представлены в следующем виде:
Восстановленные флавиннуклеотиды оксидаз L- и D-аминокислот могут непосредственно окисляться молекулярным кислородом. При этом образуется перекись водорода, которая подвергается расщеплению под действием каталазы на воду и кислород.
Помимо перечисленных 4 типов дезаминирования аминокислот и ферментов, катализирующих эти превращения, в животных тканях и печени человека открыты также три специфических фермента (серин- и треонин-дегидратазы и цистатионин-γ-лиаза), катализирующих неокислительное дезаминирование соответственно серина, треонина и цистеина.
Конечными продуктами реакции являются пируват и α-кетобутират, аммиак и сероводород. Поскольку указанные ферменты требуют присутствия пиридоксальфосфата в качестве кофермента, реакция неокислительного дезаминирования, вероятнее всего, протекает с образованием шиффовых оснований как промежуточных метаболитов.
39.обезвреживание аммиака в организме. Орнитиновый цикл образования мочевины. Аммиак – это высокотоксичное соединение, поэтому концентрация аммиака в организме должна сохраняться на низком уровне. Действительно, уровень аммиака в крови в норме не превышает 60 мкмоль/л. Аммиак должен подвергаться связыванию в тканях с образованием нетоксичных соединений, легко выделяющихся с мочой. Один из путей связывания и обезвреживания аммиака в организме, в частности в мозге, сетчатке, почках, печени и мышцах, – это биосинтез глутамина (и, возможно, аспарагина). Глутамин и аспарагин выделяются с мочой в небольшом количестве. Было высказано предположение, что они выполняют скорее транспортную функцию переноса аммиака в нетоксичной форме. Ниже приводится химическая реакция синтеза глутамина, катализируемого глутаминсинтетазой.
Часть аммиака легко связывается с α-кетоглутаровой кислотой благодаря обратимости глутаматдегидрогеназной реакции. Если учесть связывание одной молекулы аммиака при синтезе глутамина, то нетрудно видеть, что в организме имеется хорошо функционирующая система, связывающая две молекулы аммиака:
Глутамин, кроме того, используется почками в качестве резервного источника аммиака (образуется из глутамина под действием глутаминазы), необходимого для нейтрализации кислых продуктов обмена при ацидозе и защищающего тем самым организм от потери с мочой используемых для этих целей ионов Na+. Основным механизмом обезвреживания аммиака в организме является биосинтез мочевины. Последняя выводится с мочой в качестве главного конечного продукта белкового, соответственно аминокислотного, обмена. Таким образом, весь цикл мочевинообразования может быть представлен следующим образом. На первом этапе синтезируется макроэргическое соединение карбамоилфосфат – метаболически активная форма аммиака, используемая в качестве исходного продукта для синтеза пиримидиновых нуклеотидов (соответственноДНК и РНК) и аргинина (соответственно белка и мочевины):
К настоящему времени открыты три разных пути синтеза карбамоил-фосфата de novo, катализируемые тремя разными ферментами. Первую необратимую реакцию катализирует регуляторный фермент – аммиакзави-симая карбамоилфосфатсинтетаза
На втором этапе цикла мочевинообразования происходит конденсация карбамоилфосфата и орнитина с образованием цитруллина; реакцию катализирует орнитин-карбамоилтрансфераза (формула в книге страница 352). На следующей стадии цитруллин превращается в аргинин в результате двух последовательно протекающих реакций. Первая из них, энергозави-симая,– это конденсация цитруллина и аспарагиновой кислоты с образованием аргининосукцината (эту реакцию катализирует аргининосукцинат-синтетаза). Аргининосукцинат распадается в следующей реакции на аргинин и фумарат при участии другого фермента – аргининосукцинатлиазы. На последнем этапе аргинин расщепляется на мочевину и орнитин под действием аргиназы.
Рис. 12.5. Орнитиновый цикл синтеза мочевины в печени. Из приведенной схемы процесса мочевинообразования нетрудно видеть, что один из атомов азота мочевиныимеет своим источником свободный аммиак (через карбамоилфосфат); второй атом азота поступает из ас-партата. Аммиак образуется главным образом в процессе глутаматде-гидрогеназной реакции. В процессе пополнения запасов аспартата участвуют три сопряженные реакции: сначала фумарат под действиемфумаразы присоединяет воду и превращается в малат, который окисляется при участии малатдегидрогеназыс образованием оксалоацетата; последний в реакции трансаминирования с глутаматом вновь образует аспартат. Учитывая известные фактические данные о механизмах обезвреживания аммиака в организме, можно сделать следующее заключение. Часть аммиака используется на биосинтез аминокислот путем восстановительного аминирования α-кетокислот по механизму реакции трансаминирования. Аммиак связывается при биосинтезеглутамина и аспарагина. Некоторое количество аммиака выводится с мочой в виде аммонийных солей. В форме креатинина, который образуется из креатина и креатинфосфата, выделяется из организмазначительная часть азота аминокислот. Наибольшее количество аммиака расходуется на синтез мочевины, которая выводится с мочой в качестве главного конечного продукта белкового обмена в организме человека и животных. Подсчитано, что в состоянии азотистого равновесия организм взрослого здорового человека потребляет и соответственно выделяет примерно 15 г азота в сутки; из экскретируемого с мочой количестваазота на долю мочевины приходится около 85%, креатинина – около 5%, аммонийных солей – 3%, мочевой кислоты – 1% и на другие формы – около 6%. ДЕКАРБОКСИЛИРОВАНИЕ. БИОГЕННЫЕ АМИНЫ. БИОЛОГИЧЕСКОЕ ЗНАЧЕНИЕ. ПРИМЕРЫ. Процесс отщепления карбоксильной группы аминокислот в виде СО2 получил название декарбоксилирования. Несмотря на ограниченный круг субстратов, подвергающихся декарбоксилированию в животных тканях, образующиеся продукты реакции, названные биогенными аминами, оказывают сильное фармокологическое действие на множество физиологических функций человека и животных. В живых организмах открыты 4 типа декарбоксилирования: 1.альфа-декарбоксилирование. Характерное для тканей животных, при котором от АМК отщепляется карбоксильная группа, стоящая по соседству с альфа-углеродным атомом. Продуктом реакции является СО2 и биогенные амины. 2.w-декарбоксилирование, свойственное микроорганизмам. Например, из аспарагиновой кислоты этим путем образуется альфа-аланин. 3.декарбоксилирование, связанное с реакцией трансаминирования. 4.декарбоксилирование, связанное с реакцией конденсации двух молекул. Декарбоксилирование облегчается для к-т, содержащих в a-положении электроотрицательные группы. Легкое декарбоксилирование ацетоуксусной (ф-ла I) и нитроуксусной к-т (II) обусловлено возникновением циклич. переходного состояния:
|
|||||||||
|
|
Последнее изменение этой страницы: 2017-01-24; просмотров: 597; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.142.241.200 (0.014 с.) |



















 ССООН
ССООН  НС=СН + СО2. Ароматич. к-ты декарбоксилируются, как правило, в жестких условиях, напр., при нагр. в хинолине в присут. металлич. порошков. Таким методом в присут. Сu получают фуран из пирослизевой к-ты. Декарбоксилирование ароматич. к-т облегчается при наличии электроф. заместителей, напр., тринитробензойная к-та декарбоксилируется при нагр. до 40-45 °С. Декарбоксилирование паров карбоновых к-т над нагретыми катализаторами (карбонаты Са и Ва, Аl2О3 и др.) - один из методов синтеза кетонов: 2RCOOH: RCOR + Н2О + СО2. При декарбоксилировании смеси двух к-т образуется смесь несимметричного и симметричного кетонов. Декарбоксилирование натриевых солей карбоновых к-т при электролизе их конц. водных р-ров - важный метод получения алканов. К р-циям декарбоксилирования, имеющим препаративное значение, относится галогендекарбоксилирование - замещение карбоксильной группы в молекуле на галоген. Р-ция протекает под действием LiCl (или N-бромсукцинимида) и тетраацетата Рb на карбоновые к-ты, а также своб. галогенов (Сl2, Вr2, I2) на соли карбоновых к-т, напр.: RCOOM
НС=СН + СО2. Ароматич. к-ты декарбоксилируются, как правило, в жестких условиях, напр., при нагр. в хинолине в присут. металлич. порошков. Таким методом в присут. Сu получают фуран из пирослизевой к-ты. Декарбоксилирование ароматич. к-т облегчается при наличии электроф. заместителей, напр., тринитробензойная к-та декарбоксилируется при нагр. до 40-45 °С. Декарбоксилирование паров карбоновых к-т над нагретыми катализаторами (карбонаты Са и Ва, Аl2О3 и др.) - один из методов синтеза кетонов: 2RCOOH: RCOR + Н2О + СО2. При декарбоксилировании смеси двух к-т образуется смесь несимметричного и симметричного кетонов. Декарбоксилирование натриевых солей карбоновых к-т при электролизе их конц. водных р-ров - важный метод получения алканов. К р-циям декарбоксилирования, имеющим препаративное значение, относится галогендекарбоксилирование - замещение карбоксильной группы в молекуле на галоген. Р-ция протекает под действием LiCl (или N-бромсукцинимида) и тетраацетата Рb на карбоновые к-ты, а также своб. галогенов (Сl2, Вr2, I2) на соли карбоновых к-т, напр.: RCOOM  RHal (М = Ag, К, Hg, T1). Серебряные соли дикарбоновых к-т под действием I2 легко превращаются в лактоны:
RHal (М = Ag, К, Hg, T1). Серебряные соли дикарбоновых к-т под действием I2 легко превращаются в лактоны: 



