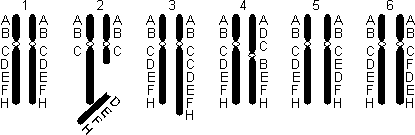Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Пердмет и методы изучения генетики. Этапы развития, роль отечетвенных учёных в развитии генетики.Стр 1 из 24Следующая ⇒
ПЕРДМЕТ И МЕТОДЫ ИЗУЧЕНИЯ ГЕНЕТИКИ. ЭТАПЫ РАЗВИТИЯ, РОЛЬ ОТЕЧЕТВЕННЫХ УЧЁНЫХ В РАЗВИТИИ ГЕНЕТИКИ. Генетика – наука, изучающая основные закономерности наследственности и изменчивости, которые относятся к основным свойствам живого. Наследственность – свойство организмов повторять в ряду поколений сходные признаки. Единица наследственности – ген, реализующийся в признак. Изменчивость – способность организмов приобретать новые признаки. Наследование – способ передачи генетической информации, которая может изменяться в зависимости от форм размножения. Методы изучения генетики: - Гибридологический - Популяционно-статистический - Цитогенетический - Онтогенетический В развитии генетики можно выделить 3 этапа: 1. (с 1900 по 1925 г.) – этап классической генетики. В этот период были переоткрыты и подтверждены на многих видах растений и животных законы Г.Менделя, создана хромосомная теория наследственности (Т.Г.Морган). 2. (с 1926 по 1953) – этап широкого развёртывания работ по искусственному мутагенезу (Г.Меллер и др.). в это время было показано сложное строение и дробимость гена, заложены основы биохимической, популяционной и эволюционной генетики, доказано, что молекула ДНК является носителем наследственной информации (О.Эвери), были заложены основы ветеринарной генетики. 3. (начинается с 1953 г.) – этап современной генетики, для которого характерны исследования явлений наследственности на молекулярном уровне. Была открыта структура ДНК (Дж. Утсон), расшифрован генетический код (Ф.Крик), химическим путём синтезирован ген (Г. Корана). Большой вклад в развитие генетики внесли отечественные учёные. Научные генетические школы созданы Вавиловым и др. Получили искусственным путём мутации – Филиппов. Вавилов сформулировал закон гомологических рядов наследственной изменчивости. Карпеченко предложил метод преодоления бесплодия у некоторых гибридов. Четвериков – основатель учения о генетике популяций. Серебровский – показал сложное строение и дробимость гена. Н.К.Кольцова по праву называют основоположником российской экспериментальной биологии. Он первым разработал гипотезу молекулярного строения и матричной репродукции хромосом, предвосхитившую принципиальные положения современной генетики.
МОНОГИБРИДНОЕ СКРЕЩИВАНИЕ. ФОРМЫ НАСЛЕДОВАНИЯ, ПРАВИЛА, ЦИТОЛОГИЧЕСКИЕ ОБОСНОВАНИЯ Моногибридное скрещивание - скрещивание форм, отличающихся друг от друга по одной паре изучаемых альтернативных признаков, за которые отвечают аллели одного гена. Закон Менделя: Закон единообразия гибридов 1 поколения При скрещивании гомозиготных особей, отличающихся друг от друга по 1 паре альтернативных признаков, всё потомство в 1 поколении единообразно как по фенотипу, так и по генотипу.
ДИ- И ПОЛИГИБРИДНОЕ СКРЕЩИВАНИЕ. НЕЗАВИСИМОЕ КОМБИНИРОВАНИЕ И ЕГО ЦИТОЛОГИЧЕСКИЕ ОСНОВЫ. МНОЖЕСВТЕННЫЕ АЛЛЕЛИ И ПОЛИГЕННОЕ НАСЛЕДОВАНИЕ НА ПРИМЕРЕ ЧЕЛОВЕКА. Множественный аллелизм — это существование в популяции более двух аллелей данного гена. В популяции оказываются не два аллельных гена, а несколько. Возникают в результате разных мутаций одного локуса. Гены множественных аллелей взаимодействуют между собой различным образом. Так, кроме основных доминантного и рецессивного аллельных генов, между ними возникают промежуточные, которые по отношению к доминатному ведут себя как рецессивные, а по отношению к рецессивному - как доминантные гены. В популяциях как гаплоидных, так и диплоидных организмов обычно существует множество аллелей, для каждого гена. Это следует из сложной структуры гена — замена любого из нуклеотидов или иные мутации приводят к появлению новых аллелей. Видимо, лишь в очень редких случаях любая мутация столь сильно влияет на работу гена, а ген оказывается столь важным, что все его мутации приводят к гибели носителей. Так, для хорошо изученных у человека глобиновых генов известно несколько сотен аллелей, лишь около десятка из них приводит к серьёзным патологиям. По типу множественных аллелей наследуются группы крови О, А, В и АВ у человека. Несколько упрощая фактическое положение вещей, можно сказать, что четыре группы крови человека определяются антигенами А и В. Если ни одного из них нет, то у человека первая (нулевая) группа крови. Присутствие антигена А дает вторую группу, антигена В - третью, совместное их присутствие обусловливает развитие четвертой группы. Сделано предположение, что нулевая группа зависит от рецессивного гена, обозначаемого через i, над ним доминирует как ген IA, дающий вторую группу, так и ген Iв, дающий третью группу. Гены IA и IB вместе дают четвертую группу крови. Первая группа крови бывает лишь при генотипе ii, вторая - при генотипах IАIА и IAi, третья - при генотипах IВIB и IBi, четвертая - при генотипе IАIВ.
Большинство количественных признаков определяется несколькими неаллельными генами (полигенами). Их взимодейсвие в процессе формирования признака – полимерное. В этом случае 2 и более полимерные аллели в гомозиготном положении в равной степени влияют на развитие одного и того же признака. Расщепление по фенотипу: 1:4:6:4:1 Если интенсивность признака зависит от числа А в генотие – кумулятивная полимерия (Пигментация кожи А-темный, а-светлый) Если на наличие признака влияет наличие хоть 1 А в генотипе – некумулятивная ГЕНОКОПИЯ (от ген и лат. copia — множество, запас), одинаковые изменения фенотипа, обусловленные аллелями разл. генов. несколькими генами. Поскольку биосинтез молекул в клетке, как правило, осуществляется многоэтапно, мутации разных генов, контролирующих соответственно разл. этапы одного биохимич. пути, могут приводить к одинаковому результату — отсутствию конечного продукта цепи реакций и, следовательно, одинаковому изменению фенотипа. Напр., известны рецессивные аллели разл. генов, к-рые локализованы в разл. хромосомах дрозофилы, но каждый из них обусловливает одну и ту же ярко-красную окраску глаз, т. к. вызывает нарушения одного из этапов синтеза коричневого пигмента. Строго говоря, изменения фенотипа в случае Г. будут отличаться друг от друга, поскольку исходные изменения касаются всё же разл. этапов биосинтеза. Так, у человека известно неск. форм рецессивной наследств, глухоты, вызываемых мутантными аллелями, по крайней мере, трёх аутосомных генов и одного гена в Х-хромосоме. Однако в разных случаях глухота сопровождается, напр., или пигментным ретинитом, или зобом, или аномальной электрокардиограммой. Проблема Г. (как и фенокопий) особенно актуальна в мед. генетике для прогноза возможного проявления наследств, заболеваний у потомков, если родители имеют сходные болезни или аномалии развития. ФЕНОКОПИЯ Фенокопии — изменения фенотипа под влиянием неблагоприятных факторов среды, по проявлению похожие на мутации. В медицине фенокопии — ненаследственные болезни, сходные с наследственными. Распространенная причина фенокопий у млекопитающих — действие на беременных тератогенов различной природы, нарушающих эмбриональное развитие плода (генотип его при этом не затрагивается). При фенокопиях изменённый под действием внешних факторов признак копирует признаки другого генотипа (например, у человека приём алкоголя во время беременности приводит к комплексу нарушений,которые до некоторой степени могут копировать симптомы болезни Дауна). Генные мутации Генные мутации — изменения структуры генов. Поскольку ген представляет собой участок молекулы ДНК, то генная мутация представляет собой изменения в нуклеотидном составе этого участка. Генные мутации могут происходить в результате: 1) замены одного или нескольких нуклеотидов на другие; 2) вставки нуклеотидов; 3) потери нуклеотидов; 4) удвоения нуклеотидов; 5) изменения порядка чередования нуклеотидов. Эти мутации приводят к изменению аминокислотного состава полипептидной цепи и, следовательно, к изменению функциональной активности белковой молекулы. Благодаря генным мутациям возникают множественные аллели одного и того же гена.
Заболевания, причиной которых являются генные мутации, называются генными (фенилкетонурия, серповидноклеточная анемия, гемофилия и т.д.). Наследование генных болезней подчиняется законам Менделя. Хромосомные мутации Это изменения структуры хромосом. Перестройки могут осуществляться как в пределах одной хромосомы — внутрихромосомные мутации (делеция, инверсия, дупликация, инсерция), так и между хромосомами — межхромосомные мутации (транслокация). Делеция — утрата участка хромосомы (2); инверсия — поворот участка хромосомы на 180° (4, 5); дупликация — удвоение одного и того же участка хромосомы (3); инсерция — перестановка участка (6).
Хромосомные мутации: 1 — пара хромосом; 2 — делеция; 3 — дупликация; 4, 5 — инверсия; 6 — инсерция.
Транслокация — перенос участка одной хромосомы или целой хромосомы на другую хромосому. Заболевания, причиной которых являются хромосомные мутации, относятся к категории хромосомных болезней. К таким заболеваниям относятся синдром «крика кошки» (46, 5р-), транслокационный вариант синдрома Дауна (46, 21 t2121) и др. Геномные мутации Геномной мутацией называется изменение числа хромосом. Геномные мутации возникают в результате нарушения нормального хода митоза или мейоза. Гаплоидия — уменьшение числа полных гаплоидных наборов хромосом. Полиплоидия — увеличение числа полных гаплоидных наборов хромосом: триплоиды (3 n), тетраплоиды (4 n) и т.д. Гетероплоидия (анеуплоидия) — некратное увеличение или уменьшение числа хромосом. Чаще всего наблюдается уменьшение или увеличение числа хромосом на одну (реже две и более). Наиболее вероятной причиной гетероплоидии является нерасхождение какой-либо пары гомологичных хромосом во время мейоза у кого-то из родителей. В этом случае одна из образовавшихся гамет содержит на одну хромосому меньше, а другая — на одну больше. Слияние таких гамет с нормальной гаплоидной гаметой при оплодотворении приводит к образованию зиготы с меньшим или большим числом хромосом по сравнению с диплоидным набором, характерным для данного вида: нулесомия (2 n - 2), моносомия (2 n - 1), трисомия (2 n + 1), тетрасомия (2 n + 2) и т.д.
На генетических схемах, приведенных ниже, показано, что рождение ребенка с синдромом Клайнфельтера или синдромом Тернера-Шерешевского можно объяснить нерасхождением половых хромосом во время анафазы 1 мейоза у матери или у отца.
Спонтанные мутации. Мутации, помимо качественных свойств, характеризует и способ возникновения. Спонтанные (случайные) – мутации, возникающие при нормальных условиях жизни. Спонтанный процесс зависит от внешних и внутренних факторов (биологические, химические, физические). Спонтанные мутации возникают у человека в соматических и генеративных тканях. Метод определения спонтанных мутаций основан на том, что у детей появляется доминантный признак, хотя у его родителей он отсутствует. Проведенное в Дании исследование показали, что примерно одна из 24000 гамет несет в себе доминантную мутацию. Ученый же Холдейн рассчитал среднюю вероятность появления спонтанных мутаций, которая оказалась равна 5*10-5 за поколение. Другой ученый Курт Браун предложил прямой метод оценки таких мутаций, а именно: число мутаций разделить на удвоенное количество обследованных индивидов. Индуцированные мутации. Индуцированный мутагенез – это искусственное получение мутаций с помощью мутагенов различной природы. Впервые способность ионизирующих излучений вызывать мутации была обнаружена Г.А. Надсоном и Г.С. Филлиповым. Затем, проводя обширные исследования, была установлена радиобиологическая зависимость мутаций. В 1927 году американским ученым Джозефом Мюллером было доказано, что частота мутаций увеличивается с увеличением дозы воздействия. В конце сороковых годов открыли существование мощных химических мутагенов, которые вызывали серьезные повреждения ДНК человека для целого ряда вирусов. Одним из примеров воздействия мутагенов на человека может служить эндомитоз – удвоение хромосом с последующим делением центромер, но без расхождения хромосом. Мутагены — химические и физические факторы, вызывающие наследственные изменения — мутации. Впервые искусственные мутации получены в 1925 году Г. А. Надсеном и Г. С. Филипповым у дрожжей действием радиоактивного излучения радия; в 1927 году Г. Мёллер получил мутации у дрозофилы действием рентгеновских лучей. Способность химических веществ вызывать мутации (действием иода на дрозофилы) открыта И. А. Рапопортом. У особей мух, развившихся из этих личинок, частота мутаций оказалась в несколько раз выше, чем у контрольных насекомых. Мутагенами могут быть различные факторы, вызывающие изменения в структуре генов, структуре и количестве хромосом. По происхождению мутагены классифицируют на эндогенные, образующиеся в процессе жизнедеятельности организма и экзогенные — все прочие факторы, в том числе и условия окружающей среды.
По природе возникновения мутагены классифицируют на физические, химические и биологические: Физические мутагены. · ионизирующее излучение; · радиоактивный распад; · ультрафиолетовое излучение; · моделированное радиоизлучение и электромагнитные поля; · чрезмерно высокая или низкая температура. Химические мутагены. · окислители и восстановители (нитраты, нитриты, активные формы кислорода); · алкилирующие агенты (например, иодацетамид); · пестициды (например гербициды, фунгициды); · некоторые пищевые добавки (например, ароматические углеводороды, цикламаты); · продукты переработки нефти; · органические растворители; · лекарственные препараты (например, цитостатики, препараты ртути, иммунодепрессанты). К химическим мутагенам условно можно отнести и ряд вирусов (мутагенным фактором вирусов являются их нуклеиновые кислоты — ДНК или РНК). Биологические мутагены. · специфические последовательности ДНК — транспозоны; · некоторые вирусы (вирус кори, краснухи, гриппа); · продукты обмена веществ (продукты окисления липидов); · антигены некоторых микроорганизмов. Канцерогенез — сложный патофизиологический процесс зарождения и развития опухоли. Изучение процесса канцерогенеза является ключевым моментом как для понимания природы опухолей, так и для поиска новых и эффективных методов лечения онкологических заболеваний. Канцерогенез — сложный многоэтапный процесс, ведущий к глубокой опухолевой реорганизации нормальных клеток организма. Из всех предложенных до ныне теорий канцерогенеза, мутационная теория заслуживает наибольшего внимания. Согласно этой теории, опухоли являются генетическими заболеваниями, патогенетическим субстратом которых является повреждение генетического материала клетки (точечные мутации, хромосомные аберрации и т. п.). Повреждение специфических участков ДНК приводит к нарушению механизмов контроля за пролиферацией и дифференцировкой клеток и в конце концов к возникновению опухоли. Генетический аппарат клеток обладает сложной системой контроля деления, роста и дифференцировки клеток. Изучены две регулирующие системы оказывающие кардинальное влияние на процесс клеточной пролиферации. Протоонкогены- это группа нормальных генов клетки, оказывающих стимулирующее влияние на процессы клеточного деления, посредством специфических продуктов их экспрессии. Превращение протоонкогена в онкоген (ген, определяющий опухолевые свойства клеток) является одним из механизмов возникновения опухолевых клеток. Это может произойти в результате мутации протоонкогена с изменением структуры специфического продукта экспрессии гена, либо же повышением уровня экспрессии протоонкогена при мутации его регулирующей последовательности (точечная мутация) или при переносе гена в активно транскрибируемую область хромосомы (хромосомные аберрации). На данный момент изучена канцерогенная активность протоонкогенов группы ras (HRAS, KRAS2). При различных онкологических заболеваниях регистрируется значительное повышение активности этих генов (рак поджелудочной железы, рак мочевого пузыря и т. д.). Также раскрыт патогенез лимфомы Беркитта, при которой активация протоонкогена MYC происходит в случае его переноса в область хромосом, где содержатся активно транскрибируемые гены иммуноглобулинов. Функции генов-супрессоров противоположны функциям протоонкогенов. Гены-супрессоры оказывают тормозящее влияние на процессы клеточного деления и выхода из дифференцировки. Доказано, что в ряде случаев инактивация генов-супрессоров с исчезновением их антагонистического влияния по отношению к протоонкогенам ведет к развитию некоторых онкологических заболеваний. Так, потеря участка хромосомы, содержащего гены-супрессоры, ведет к развитию таких заболеваний, как ретинобластома, опухоль Вильмса и др.
Таким образом, система протоонкогенов и генов-супрессоров формирует сложный механизм контроля темпов клеточного деления, роста и дифференцировки. Нарушения этого механизма возможны как под влиянием факторов внешней среды, так и в связи с геномной нестабильностью — теория, предложенная Кристофом Лингауром и Бертом Фогельштейном. Питер Дюсберг из Калифорнийского университета в Беркли утверждает, что причиной опухолевой трансформации клетки может быть анеуплоидия (изменение числа хромосом или потеря их участков), являющаяся фактором повышенной нестабильности генома. По мнению некоторых ученых, ещё одной причиной возникновения опухолей мог бы быть врождённый или приобретённый дефект систем репарации клеточной ДНК. В здоровых клетках процесс репликации (удвоения) ДНК протекает с большой точностью благодаря функционированию специальной системы исправления пострепликационных ошибок. В геноме человека изучено, по крайней мере, 6 генов, участвующих в репарации ДНК. Повреждение этих генов влечёт за собой нарушение функции всей системы репарации, и, следовательно, значительное увеличение уровня пострепликационных ошибок, то есть мутаций. Мутационная теория канцерогенеза — учение, согласно которому причиной возникновения злокачественных опухолей являются мутационные изменения генома клетки. В настоящее время эта теория является общепринятой. В подавляющем большинстве случаев злокачественные новообразования развиваются из одной опухолевой клетки, то есть имеют моноклональное происхождение. Согласно современным представлениям, мутации, которые в конце концов приводят к развитию опухоли, могут иметь место как в половых (около 5 % всех случаев), так и в соматических клетках. Мутации вызывают врожденные уродства и наследственные болезни человека. Поэтому насущной задачей является ограждение людей от действия мутагенов. Огромное значение в этом отношении имело запрещение испытаний ядерного оружия в атмосфере. Очень важно соблюдать меры защиты людей от радиации в атомной индустрии, при работе с изотопами, рентген-лучами. Определенную роль могут сыграть антимутагены – вещества, снижающие эффект действия мутагенов (цистеамин, хинакрин, некоторые сульфаниламиды, производные пропионовой и галловой кислот). ГЕННЫЕ БОЛЕЗНИ. Генные болезни – это большая группа заболеваний, возникающих в результате повреждения ДНК на уровне гена, употребляется в отношении моногенных заболеваний. Моногенные a. Аутосомно-доминантные (синдром Марфана) b. Аутосомно-рецессивные (алькаптонурия, фенилкетонурия, альбинизм, галактозамия) c. Доминантные с Х-хромосомой (Рахит, синдром Альпорта) d. Рецессивные с Х-хромосомой (Гемофилия, миопатия, ихтиоз) e. Сцепленные с У-хромосомой ХРОМОСОМНЫЕ БОЛЕЗНИ. Хромосомные болезни, или синдромы - это группа врожденных патологических состояний, проявляющихся множественными пороками развития, различающихся по своей клинической картине, часто сопровождающихся тяжелыми нарушениями психического и соматического развития. Основной дефект - различные степени интеллектуальной недостаточности, что может осложняться нарушениями зрения, слуха, опорно-двигательного аппарата, более выраженными, чем интеллектуальный дефект, расстройствами речи, эмоциональной сферы и поведения. Диагностические признаки хромосомных синдромов можно разделить на три группы: 1. неспецифические, т.е. такие, как выраженная умственная отсталость, сочетающаяся с дисплазиями, врожденными пороками развития и черепно-лицевыми аномалиями; 2. признаки, характерные для отдельных синдромов; 3. патогномоничные для конкретного синдрома, например, специфический плач при синдроме «кошачьего крика». Хромосомные заболевания не подчиняются менделеевским закономерностям передачи заболевания потомству и в большинстве случаев обнаруживаются спорадически, являясь следствием мутации в половой клетке одного из родителей. Хромосомные болезни могут быть унаследованы, если мутация имеется во всех клетках родительского организма. К механизмам, лежащим в основе геномных мутаций, относятся: 1. нерасхождение - хромосомы, которые должны были разделяться во время клеточного деления, остаются соединенными и относятся к одному полюсу; 2. «анафазное отставание» - утрата отдельной хромосомы (моносомия) может иметь место во время анафазы, когда одна хромосома может отстать от остальных; 3. полиплоидизация - в каждой клетке геном представлен более чем дважды. Факторы, повышающие риск рождения детей с хромосомными болезнями Причины возникновения хромосомных болезней до настоящего времени недостаточно изучены. Имеются экспериментальные данные о влиянии на мутационный процесс таких факторов, как: действие ионизирующих излучении, химических веществ, вирусов. Другими причинами нерасхождения хромосом могут быть: сезонность, возраст отца и матери, порядок рождения детей, прием лекарств во время беременности, гормональные нарушения, алкоголизм и др. Не исключается до определенной степени и генетическое детерминирование нерасхождения хромосом. Повторим, однако, что причины образования геномных и хромосомных мутаций на ранних стадиях развития зародыша до сих пор окончательно не раскрыты. К биологическим факторам повышения риска рождения детей с хромосомными аномалиями может быть отнесен возраст матери. Риск рождения больного ребенка особенно резко возрастает после 35 лет. Это характерно для любых хромосомных болезней, но наиболее четко наблюдается для болезни Дауна. В медико-генетическом планировании беременности особое значение уделяется двум факторам — наличию анеуплоидии по аутосомам у ребенка и возрасту матери старше 35 лет. К кариотипическим факторам риска у супружеских пар относятся: анеуплоидия (чаще в мозаичной форме), робертсоновские транслокации (слияние двух телоцентрических хромосом в области деления) кольцевые хромосомы, инверсии. Степень повышения риска зависит от типа хромосомных нарушений.
Синдром Дауна (трисомия по 21 паре хромосом) Причина: Нерасхождение 21 пары аутосом, транслокация 21 аутосомы на аутосому группы D или G. У 94% кариотип — 47 хромосом. Частота проявления синдрома увеличивается с возрастом матери. Клиника: Признаки, позволяющие диагностировать заболевание, в типичных случаях выявляются на самых ранних этапах жизни ребенка. Малый рост ребенка, маленькая круглая голова со скошенным затылком, своеобразное лицо - бедная мимика, косой разрез глаз со складкой у внутреннего угла, нос с широкой плоской переносицей, маленькие деформированные ушные раковины. Рот обычно полуоткрыт, язык толстый, неповоротливый, нижняя челюсть иногда выступает вперед. На щеках часто отмечается сухая экзема. Обнаруживается укорочение конечностей, особенно в дистальных отделах. Кисть плоская, пальцы рук широкие, короткие. В физическом развитии отстают, однако не резко, но нервно-психическое развитие замедленно (плохо развита речь). С возрастом выявляется ряд новых черт заболевания. Голос грубеет, отмечается близорукость, косоглазие, конъюнктивиты, неправильный рост зубов, кариес.Слабо развита иммунная система, инфекционные заболевания протекают крайне тяжело и в 15 раз чаще, чем у других детей. Встречается острый лейкоз. Патогенез: Патологии внутренних органов, сердечно-сосудистые дефекты. Диагностика: Клиническое обследование, подтверждаемое цитогенетическим анализом кариотипа. Лечение: Комплексная терапия, включающая правильную организацию режима, рационально построенная медико-педагогическая работа, лечебная физкультура, массаж, медикаментозное лечение.
Синдром Тернера-Шершевского (ХО) Причина: Нерасхождение половых хромосом, отсутствие одной Х-хромосомы, кариотип - 45 хромосом. Клиника: Низкий рост,непропорциональное строение тела, полная короткая шея с крыловидными кожными складками, широкая грудная клетка, Х-образное искривление коленей. Уши деморфированы, низко расположены. Отмечается неправильный рост зубов. Половой инфантилизм. Снижение умственного развития. Патогенез: В пубертатный период недоразвитие половых органов и вторичных половых признаков, поражение сосудистой системы, аномалии мочевой системы, уменьшение остроты зрения, слуха. Диагностика: У новорожденных ее установить трудно. С возрастом диагностика основывается на клинической картине и определении патологии кариотипа и полового хроматина. Лечение: Симптоматическое, направленное на увеличение роста. Для увеличения роста используются анаболические гормоны. С 13-15 лет начинают лечение эстрогенными препаратами. Полного выздоровления не наблюдается, однако лечебные мероприятия могут улучшить состояние больных.
Синдром Клайнфельтера (XXY; XYY; XYYYY; XXXY) Причина: Нерасхождение половых хромосом, вследствие чего увеличивается число X или Y хромосом в клетке, кариотип - 47 (XXY), 48 и более хромосом. Клиника: Высокий рост, отсутствие залысин на лбу, плохой рост бороды, гинекомастия, остеохондроз, бесплодие, слаборазвиты мышцы, аномалия зубов и костной системы. Больные могут демонстрировать сниженный интеллект. С увеличением X-хромосом увеличивается умственная отсталость до полной идиотии, с увеличением Y-хромосом - агрессивность. Больные с более глубокой степенью интеллектуального дефекта могут обнаруживать ряд психопатологических признаков: они мнительны, склонны к алкоголизму, способны совершать различные правонарушения. Патогенез: В пубертатном периоде обнаруживается недоразвитие первичных половых признаков. Диагностика: Основана на клинических данных, а также на определении патологического кариотипа цитогенетическим методом, что подтверждается исследованием полового хроматина в клетках. Лечение: Терапия с помощью мужских половых гормонов для увеличения потенции. Психотерапия.
Синдром Волъфа-Хиршхорна Причина: У 80 % страдающих им новорожденных цитологическую основу данного синдрома составляет деления короткого плеча 4-й хромосомы. Размеры делеции колеблются от небольших терминальных до занимающих около половины дистальной части короткого плеча. Отмечается, что большинство делеции возникает заново, около 13 % происходит, в результате транслокаций у родителей. Реже в геноме больных, помимо траснлокации, имеются и кольцевые хромосомы. Наряду с делениями хромосом, патология у новорожденных может быть обусловлена инверсиями, дупликациями, изохромосомами. Клиника: У новорожденных небольшой вес при нормальной продолжительности беременности. Также отмечаются микроцефалия, клювовидный нос, эпикант, антимонголоидный разрез глаз (опущение наружных углов глазных щелей), аномальные ушные раковины, расщелина верхней губы и неба, маленький рот, деформация стоп и др. Дети с синдромом Вольфа-Хиршхорна маложизнеспособны, как правило умирают в возрасте до одного года. Патогенез: Болезнь характеризуется многочисленными врожденными пороками развития, задержкой умственного и психомоторного развития. Диагностика: По клинической картине. Лечение: Не существует. Синдром трисомии (XXX) Причина: Нерасхождение половых хромосом в результате нарушения работы митотического веретена деления во время мейоза, кариотип — 47 хромосом. Клиника: Пузырное нерасхождение плаценты; новорожденный имеет небольшой, широкий задний родничок, недоразвитые затылочные и теменные кости черепа. Отставание в развитии на 6-7 месяцев. Низко расположены деформированные ушные раковины. Синдактилия пальцев кисти, расщелина губы и неба, гидроцефалия. Многие женщины нормально развиты, интеллект ниже среднего. Частота развития шизофреноподобных психозов увеличивается второе. Патогенез: Пороки развития внутренних органов. Диагностика: По клинической картине и цитогенетическому определению патологии кариотипа и полового хроматина. Лечение: Симптоматическое.
Синдром Эдвардса (трисомия по 18 паре хромосом) Причина: Нерасхождение аутосом на стадии гамет (иногда зигот). Лишняя хромосома в 18 паре. Кариотип 47, Е18+. Выражена зависимость частоты рождения больных детей от возраста родителей. Клиника: Пренатальное недоразвитие, слабая активность плода, нарушения строения лица (короткие глазные щели, маленькая верхняя челюсть) и костно-мышечной системы практически постоянны. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко. Грудина короткая, ядра окостенения расположены неправильно и в меньшем количестве. Спинномозговые грыжи и расщелины губ. Патогенез: Наиболее постоянны пороки сердца и крупных сосудов. Нарушения развития головного мозга, в основном гипоплазия мозжечка и мозолистого тела. Из пороков глаз чаще всего обнаруживается микроанафтольмия. Врожденное отсутствие щитовидной железы и надпочечников. Диагностика: Клинический осмотр, дерматоглифика, цитогенетическое обследование. Лечение: Отсутствует, 90% детей умирают на первом году жизни. Выжившие дети умирают от инфекционных заболеваний, чаще от пневмонии.
Синдром Патау (трисомия но 13 таре аутосом) Причина: Нерасхождение аутосом 13 пары в гаметогенезе у одного из родителей. Кариотип - 47, D13+. Клиника: Аномалии черепа и лица, окружность черепа обычно уменьшена, в ряде случаев имеется выраженная тригоноцефалия. Умеренная микроцефалия сочетается со сравнительно низким и скошенным лбом, узкими глазными щелями, запавшим предносьем с широким основанием носа, низко расположенными и деформированными ушными раковинами. Расстояние между глазными щелями часто уменьшено. На коже головы имеются дефекты скальпа овальной или круглой формы. Часто – заячья губа и волчья пасть. Аномалии костно-мышечной системы, полидактилия. Патогенез: Смертность в течение первого года жизни (90%). Основной причиной смерти детей являются тяжелые, несовместимые с жизнью пороки развития: дефекты сердечно-сосудистой и мочеполовой систем, аномалии толстой кишки, пупочная грыжа, нарушения строения глазных яблок, постоянны микроанофтальмия, дисплазия сетчатки, катаракты. Врожденные пороки сердца встречаются у 80% детей. Диагностика: Основана на клиническом, цитогенетическим исследованиях.
Синдром "кошачьего крика" Причина: Делеция короткого плеча хромосомы 5-й пары. Кариотип 46, 5р-. Клиника: Патологическое строение голосовых связок - сужение, мягкость хрящей, отечность и необычная складчатость слизистой, мяуканье кошки. Недоразвитие речи. Микроцефалия. Лунообразное лицо, монголоидный разрез глаз, косоглазие, катаракта, атрофия зрительного нерва, плоская спинка носа, высокое нёбо, деформированные ушные раковины. Косолапость. Задержка умственного и физического развития. Продолжительность жизни значительно снижена, только около 14% больных переживают возраст 10 лет. Патогенез: Порок сердца. Диагностика: Клиническое обследование с выявлением наиболее постоянного признака синдрома - "кошачий крик", дерматоглифика и цитогенетическое выявление патологии кариотипа. Лечение: Отсутствует.
Синдром Орбели Причина: Деления длинного плеча аутосомы 13. Клиника: Лоб переходит в нос, не образуя носовой вырезки. Большое расстояние между глазами. Широкая спинка носа, высокое нёбо, низко расположенные диспластичные ушные раковины, пороки развития глаз (косоглазие, катаракта). Пороки опорно-двигательного аппарата -неспецифические аномалии (косолапость, вывих тазобедренных суставов). Задержка роста и психомоторного развития; характерна глубокая олигофрения. Больные с развернутой клинической картиной синдрома погибают на первом году жизни. Патогенез: Аномальное развитие практически всех органов и систем; микроцефалия; врожденные пороки сердца и аномалии прямой кишки. Диагностика: Цитогенетическое, клиническое обследование. Лечение: Отсутствует. Синдром Мориса Причина: Мутация гена, нарушающая образование нормального белка — рецептора, делает ткани-мишени невосприимчивому гормону, направляющему их развитие по мужскому типу. Не использовав такую возможность на определенном этапе онтогенеза, организм осуществляет развитие по женскому типу. Клиника: Появляется особь с кариотипом XY, но внешне более сходна с женщиной. Такие субъекты не способны иметь потомство, так как их половые железы (семенники) недоразвиты, а их выводные протоки часто формируются по женскому типу (недоразвитая матка, влагалище). Вторичные половые признаки также характерны для женского пола. Патогенез: Недоразвитые половые органы. Диагностика: Цитогенетическое, клиническое обследование. Лечение: Гормональная терапия. ПРАВИЛО ХАРДИ-ВАЙНДБЕРГА. ЕГО СОДЕРЖАНИЕ И МАТЕМАТИЧЕСКИЕ ВЫРАЖЕНИЕ. Генофонд описывают в частотах встречаемости аллельных вариантов генов или концентрации. Генофонд популяции характеризуется: 1) Единством. Единство генофонда популяции заключается в характеристике вида, как закрытой системы, сохранять свою однородность по наследственным свойствам. 2) Генетическим полиморфизмом. Природные популяции гетерогенны, они насыщены мутациями. При отсутствии давления внешних факторов эта гетерогенность находится в определенном равновесии. 3) Динамическим равновесием генов. В популяцию входят особи как с доминантными так и рецессивными признаками, не находящимися под контролем естественного отбора. Однако, доминантная аллель не вытесняет рецессивную. Обнаруженная закономерность называется законом Харди-Вайнберга для идеальной популяции. Это п
|
|||||||||
|
|
Последнее изменение этой страницы: 2021-07-18; просмотров: 108; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.36.141 (0.166 с.) |