
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Зависимость концентрации вещества от температуры
Решение. Так как реакция 2NO2 = 2NO +O2 является реакцией второго порядка, константу скорости химической реакции рассчи-тываем по уравнению, записанному относительно времени:
Константа скорости не зависит от времени, остается примерно постоянной. Таким образом, среднее значение константы скорости Пример 2. Рассчитать время, за которое прореагирует 10% исходного вещества в реакции 2NO2 = 2NO + O2. Решение. Если к моменту времени t прореагировало 10% исход-ного вещества, то непрореагировшая часть исходного вещества к этому моменту времени составляет С = 0,9 С0 моль/л. Подстановка полученного выражения в уравнение для данной реакции [реакции второго порядка, уравнение (9)], записанного относительно времени, позволяет рассчитать время:
Пример 3. Определить энергию активации химической реакции 2NO2 = 2NO + O2, используя значение констант скоростей k 1 и k 2 при двух значениях температуры: Т 1 и Т 2 (табл. 5).
Т а б л и ц а 5 Экспериментальные данные
Решение. Энергию активации химической реакции можно опре-делить, используя значение констант скоростей k 1 и k 2 при двух значениях температуры Т 1 и Т 2, по уравнению
Пример 4. Определить температурный коэффициент Вант-Гоффа g для скорости реакции 2NO2 = 2NO + O2 в интервале температур
Решение. Температурный коэффициент Вант-Гоффа γ для скоро-сти реакции в интервале температур Т 1/ Т 2 может быть рассчитан по формуле k 1/ k 2 =
или для данной реакции k 1/ k 2 = 7,700/0,844 = γ656−592/10, отсюда γ = 1,4. 10. Физический смысл константы равновесия
Физический смысл константы равновесия химической реакции. Многие химические процессы в химико-технологических систе-мах являются двусторонними: они могут протекать как в прямом, так и в обратном направлении. Для большинства реакций скорость пря-мой реакции падает во времени, а скорость обратной реакции растет по мере накопления реагентов.
Например, взаимодействие водорода и азота имеет обратимый характер:
 . .
Скорость прямой реакции в начальный момент имеет наибольшее значение, а затем уменьшается вследствие уменьшения концентрации водорода и азота, которые расходуются на образование аммиака:
Скорость обратной реакции в начальный момент времени имеет минимальное значение, а затем увеличивается, по мере роста кон-центрации аммиака:
Через какое-то время скорости прямой и обратной реакции ста-новятся равными. Такое состояние реакционной системы называется химическим равновесием и определяется уравнением
Отсюда получаем выражение для константы равновесия химической реакции − К равн:
 . .
Константа равновесия химической реакции представляет собой отношение констант скорости прямой и обратной реакций или отно-шение произведений равновесных концентраций (активностей, парциальных давлений) продуктов реакции к произведению концентраций (активностей, парциальных давлений) исходных веществ. В случае гетерогенной реакции, например,

К равн. = с HCl∙ c NH3.
Состояние равновесия реакционной системы при данной темпе-ратуре не зависит от того, какие вещества, образующие систему, являются исходными, а какие − продуктами реакции. Концентрации (активности, парциальные давления) участников реакции, которые соответствуют ее состоянию равновесия, называют равновесными концентрациями (активностями, парциальными давле-ниями). Они взаимно связаны друг с другом уравнением реакции и законом действия масс. Значение константы равновесия химической реакции показы-вает, насколько полно при данных условиях исходные вещества пре-вратились в продукты реакции. Например, если К равн. = 10−6, то можно сделать вывод, что реакция в прямом направлении при данных условиях (T, P) не идет и про-дуктов в равновесной смеси нет. Если же К равн.=106, то из этого следует, что исходные вещества практически полностью превратились в продукты реакции при данных условиях.
Смещение равновесия. Принцип Ле-Шателье. Изменение температуры системы, находящейся в состоянии равновесия и концентрации ее составных частей приводит к нарушению равенства скоростей прямой и обратной реакций. Такое состояние продолжается недолго. Через некоторое время равенство скоростей восстанавливается. Однако новое состояние равновесия смещено относительно первоначального в направлении той реакции, которая временно протекала с большей скоростью. Такой переход от одного равновесного состояния к другому называется смещением или сдвигом равновесия. Направление, в котором происходит смещение равновесия при изменении температуры, можно определить, руководствуясь прави-лом Вант-Гоффа, согласно которому повышение температуры вызы-вает смещение равновесия в сторону реакции, идущей с поглощением теплоты, а понижение температуры − в сторону реакции, идущей с выделением теплоты. Например, если реакция взаимодействия йода и водорода:
 , Δ H o298 = 51,9 кДж , Δ H o298 = 51,9 кДж
идет с поглощением тепла, а следовательно, обратная реакция − с выделением тепла, то при повышении температуры новое состояние равновесия окажется смещенным вправо, а при понижении темпе-ратуры – влево. Так как с повышением температуры константы скорости прямой и обратной реакций k 1 и k 2 увеличиваются в различное число раз, то в новом состоянии равновесия константа К равн.= k 1/ k 2 будет иметь другое значение. Направление, в котором происходит сдвиг равновесия при изме-нении концентрации составных частей системы, можно определить исходя из следующих соображений: при увеличении концентрации исходных веществ происходит сдвиг равновесия в сторону прямой реакции, а увеличение концентрации продуктов реакции вызывает смещения равновесия в обратном направлении. Например, если в реакционной системе H2 + I2↔2HI увеличить концентрацию йода, то увеличится скорость прямой реакции, т.е. ре-акции образования HI. Концентрация же водорода уменьшится, по-скольку он расходуется на образование HI. В реакционной системе произойдет сдвиг равновесия вправо, константа равновесия при этом не изменится. К этому выводу можно прийти исходя из выражения К равн = k 1/ k 2. Константы скорости прямой и обратной реакций не зависят от концентраций, и, следовательно, их отношение сохранит своё прежнее значение. Изменение давления также может вызвать смещение равно-весия в том случае, если в процессе реакции уменьшается или увели-чивается объем системы. При определении направления, в котором происходит сдвиг равновесия при изменении давления, следует помнить, что увеличение давления смещает равновесие в сторону образования тех продуктов, которые занимают меньший объем, и наоборот, уменьшение давления способствует смещению равновесия в сторону образования продуктов, занимающих больший объем. Так как в системе N2 + + 3H2→2NH3 объем образующегося аммиака в два раза меньше объема исходных газов, то увеличение давления вызывает смещение равновесия вправо, а уменьшение давления будет способствовать разложению аммиака на водород и азот.
Направление, в котором происходит сдвиг равновесия системы, может быть определено исходя из принципа Ле-Шателье: если из-менить одно из условий состояния системы, находящейся в равно-весии, – температуру, концентрацию, давление, то в системе усили-вается процесс, стремящийся свести это изменение к минимуму, т.е. ослабить его.
11. Ионные равновесия в растворах электролитов.
Электролиты − это вещества, растворы или расплавы которых проводят электрический ток. Носителями зарядов являются поло-жительно заряженные ионы – катионы и отрицательно заряженные ионы – анионы. Заряженные частицы в растворах электролитов появляются в результате распада (диссоциации) молекул электролита на ионы, которые несут положительный или отрицательный заряд. Этот процесс называется электролитической диссоциацией и описывается уравнением AxBy ↔ xA + + yB или, конкретно, Al2(SO4)3 ↔ 2Al3+ + +3SO42−. Число положительных зарядов равно числу отрицательных, поэтому раствор в целом остается электронейтральным. Электролитической диссоциации подвержены вещества с силь-ной полярной или ионной связью (соли, кислоты, основания) в раст-ворителях с полярными молекулами. Диссоциация молекул электролита на ионы происходит за счет электростатического взаимодействия между полярными молекулами растворенного вещества и растворителя. Образовавшиеся ионы окру-жаются молекулами воды, т.е. происходит гидратация ионов. Гидратация ионов является экзотермическим процессом, т.е. проходит с выделением тепла, причем данный тепловой эффект срав-ним с тепловыми эффектами химических реакций. Взаимодействие между ионами в растворе зависит от диэлектрической проницаемости растворителя – ε, которая показывает, во сколько раз сила взаимодействия между двумя зарядами в данной среде меньше, чем в ваку-уме. Например, диэлектрическая проницаемость воды равна 81, т.е. взаимодействие между двумя зарядами в воде будет в 81 раз меньше, чем в вакууме. Поэтому чем выше диэлектрическая проницаемость растворителя, тем легче молекулы электролита распадаются на ионы. По своей способности к диссоциации электролиты условно делятся на сильные и слабые. В растворах слабых электролитов очень малая часть молекул распадается на ионы, в то время как сильные элект-ролиты диссоциируют почти полностью. К сильным электролитам относятся почти все соли, сильные кислоты (HCl, HNO3, HClO3, HClO4), гидроксиды щелочных и щелочно-земельных металлов (кроме Be(OH)2, Mg(OH)2).
К слабым электролитам относятся вода, большинство органиче-ских оснований и кислот, фенолы, аммиак, амины, угольная кислота Например, равновесие в растворе уксусной кислоты выражается следующим образом:
CH3COOH↔CH3COO- + H+.
Количественно распад молекул слабого электролита на ионы характеризуется степенью диссоциации – α (альфа):
где Степень диссоциации зависит от многих факторов, в частности от концентрации слабого электролита, поэтому для суждения о силе слабого электролита ввели понятие константы диссоциации. Константа диссоциации в соответствии с законом действия масс для реакции CH3COOH↔CH3COO− + H+ выражается уравнением
 . .
Константа диссоциации зависит от природы растворителя и от температуры. Выражение для константы диссоциации можно запи-сать, используя степень диссоциации и концентрацию слабого элект-ролита:
Если степень диссоциации α ≪ 1, то для приближенных расчетов получим
где c – концентрация слабого электролита, моль/л. Анализ этого уравнения позволяет сделать вывод о том, что с уменьшением концентрации слабого электролита степень диссоциа-ции электролита возрастает. Зная значение α, можно рассчитать равновесные концентрации ионов и недиссоциированных молекул слабого электролита в растворе:
С CH3COOH = с − α∙ с = (1 − α) с.
Сильные электролиты при растворении в воде полностью рас-падаются на ионы. Сильное взаимодействие между ионами и поляр-ными молекулами воды приводит к тому, что свойства раствора сильных электролитов значительно отличаются от свойства слабых, где межионным взаимодействием можно пренебречь. В связи с этим вместо термина «концентрация» введено понятие «активность». Активность (а) – это эффективная концентрация с учетом электростатического взаимодействия между ионами в растворе. Активность отличается от концентрации на некоторую величину γ:
а = с γ,
где с − молярная концентрация, моль/л, γ − коэффициент активности. В сильно разбавленных растворах электролитов коэффициент актив-ности стремится к единице. Выражение для констант диссоциации (ионизации) слабого электролита целесообразно писать, используя соответствующие активности:
Химически чистая вода является слабым электролитом, и не-значительная часть молекул воды диссоциирует по уравнению
H2O↔OН− + H+.
Поэтому константа диссоциации воды
Термодинамические расчеты показывают, что диссоциации под-вержена лишь очень малая часть молекул воды. Поэтому можно считать, что равновесная концентрация недиссоциированных молекул во-ды практически равна общей концентрации, т.е.
c H2O =
Следовательно,
Таким образом, по значению К w можно легко определить кон-центрацию ионов водорода при известной концентрации ОН-и на-оборот. В чистой воде с H+ = с ОН−= Для количественной характеристики реакции среды предложено использовать водородный показатель рН раствора (рН = −lgсH+). Аналогично существует и рОН раствора (pOH = − lgсОН−) соот-ветственно: рК w = рН + pOH = 14. Таким образом, в нейтральной среде рН =7, в кислой рН < 7, в щелочной рН > 7. Гидролиз солей. Гидролизом называется обменное взаимодействие некоторых солей с водой, приводящее к образованию малодиссоциированных соединений (слабых кислот, слабых оснований или сложных ионов). Гидролизу подвергаются соли, образованные: · слабой кислотой и сильным основанием − CH3COONа- CH3COOН слабая кислота, NаОН – сильное основание; KCN-НСN − слабая кислота (цианистоводородная кислота), КОН – сильное основание); · слабым основанием и сильной кислотой − NH4Cl, AgNО3; · слабой кислотой и слабым основанием − NH4CH3COO, Пример. Рассмотрим реакцию гидролиза:
NH4Cl +H2О↔NH4ОН + НCl NH4+ + Cl−+ H2О↔NH4ОН+ Н++Cl−
Сокращенное ионно-молекулярное уравнение реакции гидролиза:
NH4+ + H2О↔NH4ОН+ Н+
Наличие Н+ в правой части уравнения говорит о том, что реакция среды кислая, т.е. рН < 7. Процесс гидролиза характеризуется кон-стантой гидролиза К г. В случае слабой кислоты
где β − степень гидролиза соли; с − концентрация раствора соли, моль/л; В случае слабого основания
Если соль образована слабой кислотой и слабым основанием,
Примеры решения типовых задач
Пример 1. В каком направлении произойдет смещение равно-весия в системе H2 +S ⇄ H2S если увеличить концентрацию водорода? Решение. Если увеличить концентрацию водорода, то увеличится скорость прямой реакции и произойдет сдвиг равновесия вправо, но константа равновесия при этом не изменится. Пример 2. В каком направлении произойдет смещение равнове-сия при повышении давления в системе 2NO + O2 ⇄ 2NO2? Решение. При повышении давления равновесие смещается в сто-рону тех реагентов, которые занимают меньший объем (т.е. в сторону реагентов – исходных или конечных, где меньше общее число моль газообразных веществ). В нашем случае равновесие смещается впра-во, так как там общее число молей равно двум, а слева – трем. Пример 3. В каком направлении произойдет смещение равно-весия при повышении температуры системы COCl2⇄CO+Cl2? Δ H 0298= = 113 кДж? Решение. Прямая реакция протекает с поглощением тепла, по-этому равновесие будет смещено вправо, так как в этом случае реакционная система уменьшает внешнее воздействие температуры. Пример 4. В какую сторону сместится равновесие реакции Решение. Так как температурные коэффициенты прямой и об-ратной реакций не равны, повышение температуры по-разному ска-жется на изменении скоростей этих реакций. При повышении тем-пературы на 30 °С скорость прямой реакции
скорость обратной реакции
Таким образом, при повышении температуры скорость прямой реакции возросла в 15,6 раза, а обратной – в 32,8 раза. Следовательно, равновесие сместится в сторону обратной реакции. Пример 5. Как изменится равновесие реакции
если увеличить давление в реакционной системе в два раза? Решение. До увеличения давления в системе выражения для скоростей прямой и обратной реакции будут следующими:
для прямой реакции для обратной реакции При увеличении давления в два раза концентрации всех реаги-рующих веществ увеличились в два раза, так как общий объем си-стемы уменьшился в два раза. Тогда скорости прямой и обратной реакций станут равными:
В результате увеличения давления скорости прямой и обратной реакций увеличились соответственно в восемь раз и четыре раза:
Таким образом, скорость прямой реакции будет в два раза больше, чем скорость обратной реакции. Следовательно, смещение равнове-сия произойдет слева направо, т.е. в сторону образования NО2. Пример 6. Определить концентрацию гидроксид-ионов и рН в 0,01 М растворе гидроксида аммония. Решение. Значение константы диссоциации NH4ОН Кд=1,77∙10−5. Электролитическая диссоциация гидроксида аммония описывается уравнением NH4ОН↔NH4++ ОН−.
Так как имеем разбавленный раствор слабого электролита, то закон разбавления Оствальда можно использовать в виде
тогда концентрация гидроксид-ионов:
или
рОН = −lg[ О H −]= −lg(4,2∙10−4) = 3,38; рН = 14 − рОН = 14 − 3,38 = 10,62.
12. Электродные потенциалы, гальванические элементы.
Электрохимия является разделом физической химии, в котором изучают закономерности процессов, протекающих при взаимном превращении электрической и химической форм энергии. В основе электрохимических процессов лежат окислительно-вос-становительные реакции, при протекании которых происходит одновременное окисление восстановителя и восстановление окислителя за счет перераспределения электронов:
Red1 + Ox2® Ox1 + Red2 Red1® Ox1 + nē – окисление Ox2 + nē ® Red2 – восстановление,
где Red1 – восстановленная, а Ox1 – окисленная форма вещества 1, Red2, Ox2 − соответственно вещества 2 (Red – от reduction – вос-становление; Ox – от oxidation – окисление). Процессы окисления и восстановления могут быть разделены в пространстве. В этом случае в реакционной системе может осущест-вляться перенос электронов между ее частями, т.е. будет протекать электрический ток. Если окислительный и восстановительный процессы разделены в пространстве, то переход электронов от восстановителя к окислителю в самопроизвольном процессе (D r G < 0) может быть использован для получения электрической энергии. При использовании ее внешнего источника можно осуществлять вынужденные химические реакции (D r G > 0). Таким образом, происходит непосредственное взаимное превращение химической и электрической форм энергии. Такие окис-лительно-восстановительные процессы называются электрохимиче-скими и происходят в электрохимической системе (ЭХС) − рис. 6. Электрод, на поверхности которого происходит реакция окисления, называется анодом, а на поверхности которого происходит реакция восстановления – катодом:
М1 x ® M1 x + n + nē – анодный процесс (Red1® Ox1 + nē), М2 x + nē ® M2 x − n – катодный процесс (Ox2 + nē ® Red2).
Электрохимический процесс сопровождается переносом зарядов. Во внешней цепи электроны движутся от анода к катоду, а во внут-ренней происходит переход ионов.
Рис. 6. Электрохимическая система (ЭХС) включает в себя электроды 1 – проводники с электронным типом проводимости (чаще всего металл), находящиеся в контакте с электролитом; электролиты 2 – проводники с ионным типом проводимости (раствор веществ, диссоциирующих на ионы); внешнюю цепь 3 – проводники, обеспечивающие возможность перехода электронов между электродами (протекания электрического тока); внутреннюю цепь 4 – проводник с ионным типом проводимости, обеспечивающий возможность перехода ионов; проводник электрического тока или внешний источник электрической энергии − 5
ЭХС принято изображать в виде условной схемы: слева записы-вается анод с электролитом (А), справа катод с электролитом (К); граница раздела «электрод–электролит» обозначается вертикальной линией; граница раздела между анодным и катодным электролитами, обеспечивающая возможность перехода ионов (ионный проводник), – двумя вертикальными параллельными линиями. Пример. Анод – цинк, находящийся в контакте с раствором суль-фата цинка; катод – медь, находящаяся в контакте с сульфатом меди:
(а)(−)Zn|ZnSO4||CuSO4|Cu(к)(+), или (а)(−)Zn|Zn+2||Cu+2|Cu(к)(+).
Если ЭХС находится в состоянии равновесия (D G = 0), то ток в цепи равен нулю. Во внешней цепи не происходит перенос элект-ронов и соответственно отсутствует направленное движение ионов во внутренней цепи. Если в ЭХС протекает ток, то она выходит из состояния рав-новесия и электрохимический процесс становится неравновесным. Он является самопроизвольным, если D G < 0. Если D G > 0, то протекание вынужденного процесса обеспечивается включением во внешнюю цепь источника электрической энергии. При самопроизвольном процессе в результате протекания реак-ций на поверхности электродов энергия химических связей превра-щается в электрическую. В этом случае электрохимическая система называется гальваническим элементом, который может служить химическим источником тока (ХИТ). При осуществлении вынужденного электрохимического процесса за счет внешнего источника электрической энергии на поверхности электродов протекают реакции превращения веществ. При этом электрическая энергия переходит в энергию новых химических связей. Химические процессы, осуществляемые за счет электрической энергии, называются электролизом, а электрохимическая система – электролизером. Изменение энергии Гиббса в электрохимическом процессе равно ее изменению в результате протекания химической реакции (D r G) плюс работа переноса электрических зарядов между электродами (W э), имеющими разность потенциалов (электродвижущую силу) Е, (E = ∆φ = jк – jа):
D G = D r G + W э ® D G = D r G + n × F × E,
где F – число Фарадея. В состоянии равновесия D G = 0. Следовательно, разность по-тенциалов электродов равна: При записи электрохимических процессов принято обозначать электроды с относительно большим потенциалом знаком «+», а с относительно меньшим потенциалом − знаком «-». Таким образом, в гальваническом элементе потенциал анода будет отрицательным, а катода «положительным. В электролизере наоборот − анод будет «+», катод «-». Скачок потенциала на границе «металл–электролит» возникает в системе, состоящей из металлического электрода, погруженного в раствор, содержащий катионы того же металла (раствор соли). Если металл находится в контакте, например, с водным раствором элект-ролита, то на границе раздела фаз «металл–электролит» протекают следующие процессы. 1. В результате физико-химического взаимодействия между ди-полями воды и поверхностными ионами металла происходит его растворение. Особенности химической связи в металлах приводят к тому, что в раствор переходят только ионы металла, а электроны ос-таются в кристалле. В результате электрод приобретает избыточный отрицательный, а раствор – избыточный положительный заряды. Этот процесс можно записать как реакцию окисления металла:
Me0 + x H2O®Me n +(H2O) x + n ē.
2. Наличие катионов металла в растворе и отрицательный заряд электрода за счет избыточных электронов обусловливает протекание обратного процесса: катионы металла переходят из раствора на по-верхность электрода. При этом они теряют свою гидратную оболочку и встраиваются в кристаллическую решетку, что приводит к умень-шению величин отрицательного заряда электрода и положительного заряда раствора. Этот процесс можно записать как реакцию восстановления металла:
Me n +(H2O) x + n ē ®Me0+ x H2O.
В результате протекания этих процессов система придет в со-стояние равновесия. Скорости первого и второго процессов будут равны: Me n +(H2O) x + n ē ↔ Me0+ x H2O или Me n ++ n ē ↔ Me0.
При этом потенциал электрода и концентрация катионов в раст-воре сохранят постоянное равновесное значение. На границе «электрод–электролит» возникает обменный двойной электрический слой, образованный с одной стороны избыточными электронами в электроде, а с другой – избыточными положительными ионами металла в растворе (противоионами). Он рассматривается как единая электронейтральная система: заряд электрода равен сумме зарядов противоионов. Поскольку существует разделение зарядов в пространстве, то между металлом и раствором возникает разность потенциалов − j. Говорят, что при контакте металла с электролитом на границе раздела возникает скачок электрического потенциала – электродный потенциал. Способов экспериментального определения и расчета абсолют-ных величин электродных потенциалов не существует. С достаточной степенью точности можно измерить разность потенциалов Dj между двумя электродами в ЭХС. Если равновесие в такой системе до-стигнуто в стандартных условиях: активность (концентрация) потенциалопределяющих ионов 1 моль/л; температура Т 0 = 298 К; давление газов р 0 = 1,013×105 Па, то измеряемая Dj равна разности между стандартными электродными потенциалами: Dj = j01-j02. Если один из электродов принять в качестве электрода срав-нения, стандартный потенциал которого условно считать равным нулю, то измеренная разность потенциалов будет являться потенциалом второго электрода относительно первого. В качестве электрода сравнения используют стандартный водородный электрод, потенциал которого условно принят равным нулю (φ02Н/2Н+ = 0). В рассматриваемой паре электродов потенциал стандартного во-дородного электрода может быть как больше, так и меньше стан-дартного потенциала измеряемого. Например, для металлических электродов электрохимические системы могут быть записаны как
(–)Me0|Men+||2H+|H20, Pt(+) или (–)H20, Pt |2H+||Men+|Me0(+).
Таким образом, потенциал измеряемого электрода относительно водородного может быть отрицательным: Измеренные относительно водородного электрода величины стандартных электродных потенциалов сведены в таблицу в порядке их возрастания. Такую последовательность называют рядом стан-дартных электродных потенциалов (табл. 6). Т а б л и ц а 6 Ряд стандартных электродных потенциалов металлов (В)
Для определения величины электродного потенциала в нестан-дартных условиях используют уравнение Нернста (9):
где R – универсальная газовая постоянная (R = 8,314 Дж/моль∙К); F – постоянная Фарадея, 96485 Кл/моль; Т − температура, К; n − число электронов, участвующих в электродном процессе; Из уравнения (9) следует, что потенциал электрода при данной температуре определяется стандартным электродным потенциалом (j0) и концентрацией (активностью) ионов, участвующих в реакции. Для стандартной температуры (T = 298 K) уравнение (9) имеет вид
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2021-05-27; просмотров: 90; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 13.59.80.187 (0.195 с.) |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 (конц.−1 время−1), (9)
(конц.−1 время−1), (9)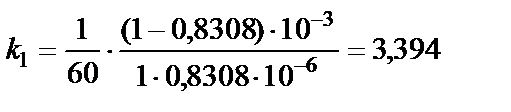 с−1∙моль−1∙л,
с−1∙моль−1∙л, с−1∙моль−1∙л,
с−1∙моль−1∙л,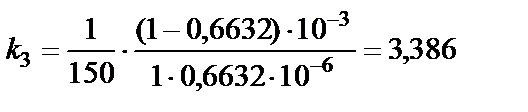 с−1∙моль−1∙л,
с−1∙моль−1∙л, с−1∙моль−1∙л,
с−1∙моль−1∙л,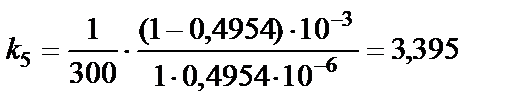 с−1∙моль−1∙л.
с−1∙моль−1∙л.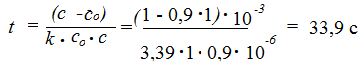 .
.

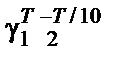


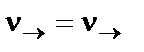 или
или 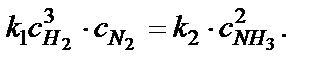
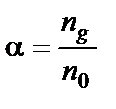 ,
, − число молекул, распавшихся на ионы, n 0 − общее число молекул растворенного вещества в растворе.
− число молекул, распавшихся на ионы, n 0 − общее число молекул растворенного вещества в растворе.
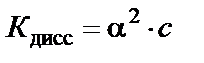 или
или 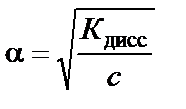 ,
, ,
,

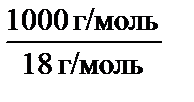 = 55,56 моль/л.
= 55,56 моль/л. ∙ c H2O =
∙ c H2O =  ∙55,56 = К w, К w – константа, которая называется ионным произведением воды. При t = 25 °С К w = 10−14. Это означает, что если c H+ = 10−2 моль/л, то
∙55,56 = К w, К w – константа, которая называется ионным произведением воды. При t = 25 °С К w = 10−14. Это означает, что если c H+ = 10−2 моль/л, то моль/л.
моль/л.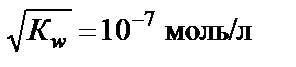 .
.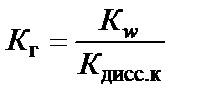 ,
,  ,
, − ионное произведение воды.
− ионное произведение воды.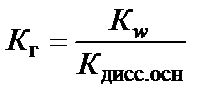 .
.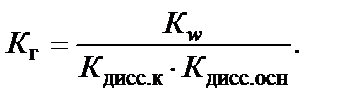

 .
. ,
,

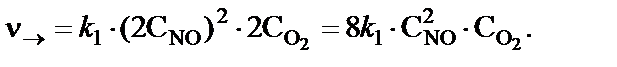

 , откуда степень диссоциации NH4ОН
, откуда степень диссоциации NH4ОН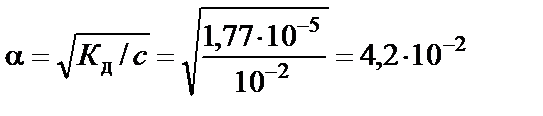 ,
,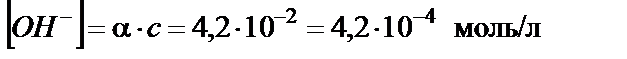
 . Термодинамическое условие самопроизвольного протекания процесса – D r G < 0. Следовательно, при работе гальванического элемента E > 0 (так как n и F больше нуля). Если потенциал катода составляет величину jк, а потенциал анода jа, то E = jк – jа > 0 и jк > jа. В случае вынужденного процесса D r G > 0. Следовательно, при электролизе разность потенциалов электродов E < 0 и соответственно jк < jа.
. Термодинамическое условие самопроизвольного протекания процесса – D r G < 0. Следовательно, при работе гальванического элемента E > 0 (так как n и F больше нуля). Если потенциал катода составляет величину jк, а потенциал анода jа, то E = jк – jа > 0 и jк > jа. В случае вынужденного процесса D r G > 0. Следовательно, при электролизе разность потенциалов электродов E < 0 и соответственно jк < jа.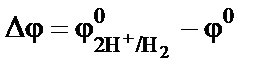 или поло-жительным:
или поло-жительным:  . За стандартный электродный потен-циал принимается измеренная разность потенциалов, соответственно
. За стандартный электродный потен-циал принимается измеренная разность потенциалов, соответственно  , (9)
, (9) – концент-рация (активность) раствора электролита (ионов металла); φ0 – стан-дартный потенциал электрода, В.
– концент-рация (активность) раствора электролита (ионов металла); φ0 – стан-дартный потенциал электрода, В. .
.


