Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Оптимизация отдельной МолекулыСодержание книги
Поиск на нашем сайте
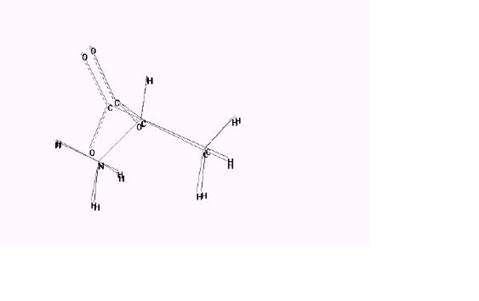
Редактирование Структуры Эта структура - только углеводород, и Вы должны редактировать структуру, чтобы создать цвиттерион аланина. Определенно, Вы должны изменить некоторые атомы на азот или кислород и сопряженную карбоксильную группу. Редактировать структуру: 1. Открыть ячейку диалога Default Element и изменить скрытый элемент на азот. 2. L-щелчок на одном из углеродов метила, чтобы превратить его в атом азота, как показано ниже: 3. Изменить скрытый элемент на кислород. 4. Сделать L-щелчок на двух водородах, которые станут частью карбоксильной группы, чтобы превратить их в атомы кислорода. 5. Сделать двойной щелчок на каждой из связей кислород-углерод, чтобы повернуть их в сопряженные связи. 6. Ваш рисунок должен напомнить это: Назначить Заряды у атомов Следующий шаг должен разместить формальные заряды на некоторые атомы, чтобы приблизительно представить распределение заряда в цвиттерионе. Вы можете получить заряды атома с HYPERCHEM, используя следующие методы: • если молекула сформирована из шаблона, типа PDB файла, шаблон, мог бы содержать заряды • выполняя квантово-механическое вычисление • делая аппроксимацию (приближение) заряда В этом упражнении, Вы сделаете простую аппроксимацию из формального заряда + 1.0 на азотном атоме и заряды по -0.5 на кислородах. Назначать заряды атома: 1. Войти в форму выбора, и установить отобранный уровень к атомам. 2. Удостоверитесь, что Multiple Selections - отмечено. 3. Выберите атом азота. 4. Выбрать Set Charge на меню Build, и назначить заряд 1.0. 5. Сделать R-щелчок в пустой области, чтобы очистить выбор. 6. Выбрать атомы кислорода, и назначить заряд -0.5. Это устанавливает заряд -0.5 для каждого из атомов кислорода. 7. Сделать R-щелчок в пустой области, чтобы отменять выбор атома. Выбор Силового поля Следующий шаг – Вы должны выбрать силовое поле. Вы должны выбрать силовое поле прежде, чем Вы вызываете Model Builder, потому что Model Builder назначает типы атома на каждый атом согласно силовому полю, которое Вы определяете. Альтернативно, Вы можете назначать типы атома в любое время, используя Calculate Types на меню Build. Однако, самый простой путь состоит в том, чтобы назначить правильное силовое поле прежде, чем Вы вызываете Model Builder. Выбирать силовое поле: 1. Выбрать Molecular Mechanics на меню Setup. Открывается диалоговая ячейка Силового поля Molecular Mechanics. 2. Выбрать AMBER, затем L-щелчок на Options. Открывается ячейка диалога Force Field Options. 3. Использовать имеющиеся скрытые значения, затем нажмите OK. Если не использовать отсечки и диэлектрическую константу, то эти значения стандартны для силового поля AMBER. 4. Нажать OK для закрытия ячейки диалога Force Field Options. 5. Нажать OK для закрытия ячейки диалога Силовое поле Molecular Mechanics. 6. Если появляется следующая диалоговая ячейка, нажать OK. Строительство и Исследование 3-D Структуры
Строить и ориентировать структуру: 1. Сделать двойной щелчок на Selection tool, чтобы вызвать Model Builder. HYPERCHEM строит и показывает первую аппроксимацию (приближение) цвиттериона аланина. 2. Вращать и перемещать структуру, чтобы посмотреть на конформацию. Сформированная модельная структура - только аппроксимация (приближение) структуры цвиттериона аланина. 3. Открыть ячейку диалога Labels на меню Display, и выбрать для атомов Chirality. Если центральный углерод - маркированный R прежде чем S, то переключитесь на инструмент рисунка, удерживая в нажатом положении клавишу [Shift], и щелкните на центральном углероде. Вы можете использовать [Shift] + L-щелчок с инструментом рисунка, чтобы вращать два соседних атом, таким образом, происходит хиральное изменение. Вы можете также выводить клиновидные связи, чтобы определить конформацию или хиральность. 4. Изменить обозначения атома снова на Symbol, и переключить назад на инструмент выбора (если Вы изменили его на инструмент Рисунка в последнем шаге). 5. Сохранить структуру как alaz.hin. Измерять свойства структуры: 1. Войти в форму выбора и сделать Л-перемещение от атома кислорода до другого атома кислорода. Model Builder строил O-C '-O угол в 120 градусов. 2. R-щелчок в пустой области, чтобы очистить выбор. 3. Сделать Л-перемещение от атома азота до одного из атомов кислорода. N-Cα -C '-O торзионный угол ± 60 градусов или ± 120 градусов, в зависимости от порядка, который Вы применили при выборе атомов в структуре. 4. Сделать R-щелчок в пустой области, чтобы отменять выбор торзионного угла. Показывать типы атомов AMBER: 1. Открыть ячейку диалога Label, и установить тип обозначения атома на Type. Выполнение Single Point Вычисления. Теперь выполните одноточечное вычисление, чтобы вычислить энергию и градиент. Выполнять одноточечное вычисление: 1. Выбрать Single Point на меню Compute. Через некоторое время, вычисление заканчивается, и линия состояния сообщает о результатах: Энергия = 74.59 Градиент = 95.70. Сохранение Структуры Вы должны сохранить оптимизированную изолированную структуру аланина так, чтобы Вы могли сравнивать её с сольватированной структурой, которую Вы создаете позднее. Сохранять структуру: 1. Открыть ячейку диалога Save в меню File, нажимая [Ctrl] + [S] как альтернативный выбор меню. 2. Удостоверитесь, что вы бран формат(расширение) файла как HIN. 3. Дайте имя файлу ala-gas.hin. 4. Сделать L-щелчок в ячейке Comments. Как показано в следующей иллюстрации, и войдите в комментарии для файла: 5. Выбрать OK. Удаление Молекул воды Прежде чем сопоставлять, удаляют молекулы воды из изолированной структуры. Удалять молекулы воды: 1. Войти в форму выбора, и установить выбранный уровень Molecules. 2. Сделать L-щелчок на аланине. Это выбирает аланин. 3. Выбрать Complement Selection. Это выбирает только молекулы воды. 4. Выбрать Clear на меню Edit. 5. Если появляется ячейка диалога, спрашивая, хотите Вы удалить отобранные атомы, выбрать Yes. 6. Убрать метку с Show Periodic box и показывается только молекула аланина. 7. Сохранить файл как ala-sol.hin. Соединение Двух Систем Следующий шаг должен слить эту систему с изолированной системой. Прежде, чем Вы соединить две системы, Вы должны выбрать текущую систему так, чтобы Вы могли дифференцировать её на рабочем пространстве после слияния. Соединить две системы: 1. Выбрать аланин. 2. Выбрать Name Selection на меню Select, и сохранить выбор как solvated. Это удобно,чтобы выбрать его позже, если Вы захотите восстановить его. 3. Выбрать Merge на меню File, и открыть ala-gas.hin. И изолированная и сольватированная структуры должны теперь появиться на экране. Сольватированная структура должна все еще быть выбраной. Merge позволяет Вам соединять текущую систему и другую систему, которая сохранена в файле. Этот союз становится текущей системой. 4. Если необходимо, используйте инструмент Translation, с правой кнопкой мыши, пока обе структуры не будут полностью представлены. Используя обозначения и цвет, укажите, как различать молекулы. 5. Окрасить выбранную сольватированную структуру в желтый цвет и маркировать молекулу символом. Обратите внимание: Если не используется как выбор Толстая линия Selection в ячейке диалога Preferences, сольватированная структура не видна желтой, пока его не отменят. 6. Выбрать Complement Selection на меню Select. Сольватированная система отменяется, и изолированный аланин выбран. 7. Окрасить выбранный изолированный аланина фиолетовым цветом, и маркировать его символом. 8. Сделать R-щелчок в пустой области, чтобы отменять выбор изолированного аланина. Сопоставьте молекулы. Когда система включает, по крайней мере, две молекулы, как в этом примере, HYPERCHEM может сопоставлять молекулы. Суперпозиция основана на выборе трех коллинеарных атомов в каждой молекуле. Чтобы начинать сопоставление, надо первые выбранные атомы каждой молекулы сделать совпадающими. Затем, связи между первыми и вторыми выбранными атомами каждой молекулы сделать коллинеарными. Наконец, выбрать третьи атомы каждой молекулы, и поместить в ту же самую плоскость. Выполнять суперпозицию: 1. Войти в форму выбора, установите отобранный уровень на Аtoms, и пометьте ۷ Multiple Selections 2. Сделайте Л-перемещение по углу связи N-Cα -C' каждой молекулы. Вам может быть придется вращать структуры, чтобы сделать это. 3. Выбрать Overlay на меню Display. 4. Сделать R-щелчок в пустой области, чтобы очистить выбор. 5. Нажать участок пространства, чтобы сосредоточить наложенные структуры в рабочем пространстве. Текущая система должна теперь выглядеть подобной этой: 6. Выбрать Save As на меню File, и сохранить суперналоженные молекулы как ala-sup.hin. Продолжение Динамики 1. Выбрать OK, чтобы возвратиться к ячейке диалога Molecular Dynamics Options, затем выбрать, Proceed, чтобы начать динамику. Колонка под названием, Molecular Dynamics Results открывается на рабочем пространстве. 2. Переместить колонку так, чтобы Вы могли наблюдать моделирование. В то время когда выполняется вычисление, Вы можете изменять вид системы, используя инструменты вращения, перемещения, увеличения, и отрезания. Вы можете также использовать приложения в другом окне, но учтите что это может замедлить моделирование. 3. Пока продолжается моделирование, выбрать Rescale, чтобы повторно масштабировать подготавливаемые значения. Как только фаза нагревания закончена, (когда энергия увеличивается) полная энергия остается постоянной, а кинетической энергия зеркальна потенциальной энергии. Предостережение: Вы не можете восстановить график, если Вы сделаете L-щелчок на Done. Чтобы восстановить график, Вы должны использовать replay динамику.
Когда вычисление динамики закончится, Вы можете оптимизировать систему, чтобы определить новый minimum. После того, как пройдет приблизительно 10 минут, движение заканчиваются. Редактирование Структуры Эта структура - только углеводород, и Вы должны редактировать структуру, чтобы создать цвиттерион аланина. Определенно, Вы должны изменить некоторые атомы на азот или кислород и сопряженную карбоксильную группу. Редактировать структуру: 1. Открыть ячейку диалога Default Element и изменить скрытый элемент на азот. 2. L-щелчок на одном из углеродов метила, чтобы превратить его в атом азота, как показано ниже: 3. Изменить скрытый элемент на кислород. 4. Сделать L-щелчок на двух водородах, которые станут частью карбоксильной группы, чтобы превратить их в атомы кислорода. 5. Сделать двойной щелчок на каждой из связей кислород-углерод, чтобы повернуть их в сопряженные связи. 6. Ваш рисунок должен напомнить это: Назначить Заряды у атомов Следующий шаг должен разместить формальные заряды на некоторые атомы, чтобы приблизительно представить распределение заряда в цвиттерионе. Вы можете получить заряды атома с HYPERCHEM, используя следующие методы: • если молекула сформирована из шаблона, типа PDB файла, шаблон, мог бы содержать заряды • выполняя квантово-механическое вычисление • делая аппроксимацию (приближение) заряда В этом упражнении, Вы сделаете простую аппроксимацию из формального заряда + 1.0 на азотном атоме и заряды по -0.5 на кислородах. Назначать заряды атома: 1. Войти в форму выбора, и установить отобранный уровень к атомам. 2. Удостоверитесь, что Multiple Selections - отмечено. 3. Выберите атом азота. 4. Выбрать Set Charge на меню Build, и назначить заряд 1.0. 5. Сделать R-щелчок в пустой области, чтобы очистить выбор. 6. Выбрать атомы кислорода, и назначить заряд -0.5. Это устанавливает заряд -0.5 для каждого из атомов кислорода. 7. Сделать R-щелчок в пустой области, чтобы отменять выбор атома. Выбор Силового поля Следующий шаг – Вы должны выбрать силовое поле. Вы должны выбрать силовое поле прежде, чем Вы вызываете Model Builder, потому что Model Builder назначает типы атома на каждый атом согласно силовому полю, которое Вы определяете. Альтернативно, Вы можете назначать типы атома в любое время, используя Calculate Types на меню Build. Однако, самый простой путь состоит в том, чтобы назначить правильное силовое поле прежде, чем Вы вызываете Model Builder. Выбирать силовое поле: 1. Выбрать Molecular Mechanics на меню Setup. Открывается диалоговая ячейка Силового поля Molecular Mechanics. 2. Выбрать AMBER, затем L-щелчок на Options. Открывается ячейка диалога Force Field Options. 3. Использовать имеющиеся скрытые значения, затем нажмите OK. Если не использовать отсечки и диэлектрическую константу, то эти значения стандартны для силового поля AMBER. 4. Нажать OK для закрытия ячейки диалога Force Field Options. 5. Нажать OK для закрытия ячейки диалога Силовое поле Molecular Mechanics. 6. Если появляется следующая диалоговая ячейка, нажать OK. Строительство и Исследование 3-D Структуры
Строить и ориентировать структуру: 1. Сделать двойной щелчок на Selection tool, чтобы вызвать Model Builder. HYPERCHEM строит и показывает первую аппроксимацию (приближение) цвиттериона аланина. 2. Вращать и перемещать структуру, чтобы посмотреть на конформацию. Сформированная модельная структура - только аппроксимация (приближение) структуры цвиттериона аланина. 3. Открыть ячейку диалога Labels на меню Display, и выбрать для атомов Chirality. Если центральный углерод - маркированный R прежде чем S, то переключитесь на инструмент рисунка, удерживая в нажатом положении клавишу [Shift], и щелкните на центральном углероде. Вы можете использовать [Shift] + L-щелчок с инструментом рисунка, чтобы вращать два соседних атом, таким образом, происходит хиральное изменение. Вы можете также выводить клиновидные связи, чтобы определить конформацию или хиральность. 4. Изменить обозначения атома снова на Symbol, и переключить назад на инструмент выбора (если Вы изменили его на инструмент Рисунка в последнем шаге). 5. Сохранить структуру как alaz.hin. Измерять свойства структуры: 1. Войти в форму выбора и сделать Л-перемещение от атома кислорода до другого атома кислорода. Model Builder строил O-C '-O угол в 120 градусов. 2. R-щелчок в пустой области, чтобы очистить выбор. 3. Сделать Л-перемещение от атома азота до одного из атомов кислорода. N-Cα -C '-O торзионный угол ± 60 градусов или ± 120 градусов, в зависимости от порядка, который Вы применили при выборе атомов в структуре. 4. Сделать R-щелчок в пустой области, чтобы отменять выбор торзионного угла. Показывать типы атомов AMBER: 1. Открыть ячейку диалога Label, и установить тип обозначения атома на Type. Выполнение Single Point Вычисления. Теперь выполните одноточечное вычисление, чтобы вычислить энергию и градиент. Выполнять одноточечное вычисление: 1. Выбрать Single Point на меню Compute. Через некоторое время, вычисление заканчивается, и линия состояния сообщает о результатах: Энергия = 74.59 Градиент = 95.70. Оптимизация отдельной Молекулы Большая энергия и градиент указывает, что сформированная модельная структура далека от оптимальной. Основное напряжение создается из-за C-N и C-O расстояний. Оптимизировать структуру: 1. Выбрать Geometry Optimization на меню Compute. Открывается ячейка диалога Optimization Molecular Mechanics: 2. Заполнить ячейку диалога с показанными значениями, и нажать OK, Через некоторое время, минимизация закончена. Линия состояния показывает следующие значения: Энергия = 18.10 Градиент = 0.10 3. Когда оптимизация выполнена, снова измерить угол O-C '- O- торзионный угол - и N-Cα -C '-O: O-C '-O угол = 123.9 градусов N-Cα -C '-O скручивание = -51 и 99 градусов Известно, что для неоптимизированной структуры, измеренный угол O-C '-O 120 градусов, а торзионные углы N-Cα -C '-O, имеют значения ±60 градусов и ± 120 градусов. Позже в этом уроке, Вы используете N-Cα -C '-O угол, чтобы демонстрировать особенности молекулярной динамики HYPERCHEM, поэтому сохраните этот угол под каким либо названием. С все еще выбранным углом, выберите Name Selection на меню Select. Выберите - Other и напишите ncco, затем нажмите OK. 4. Сделать R-щелчок в пустой области, чтобы отменить выбранный угол. Сохранение Структуры Вы должны сохранить оптимизированную изолированную структуру аланина так, чтобы Вы могли сравнивать её с сольватированной структурой, которую Вы создаете позднее. Сохранять структуру: 1. Открыть ячейку диалога Save в меню File, нажимая [Ctrl] + [S] как альтернативный выбор меню. 2. Удостоверитесь, что вы бран формат(расширение) файла как HIN. 3. Дайте имя файлу ala-gas.hin. 4. Сделать L-щелчок в ячейке Comments. Как показано в следующей иллюстрации, и войдите в комментарии для файла: 5. Выбрать OK.
|
||||
|
|
Последнее изменение этой страницы: 2017-02-07; просмотров: 308; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.220 (0.008 с.) |