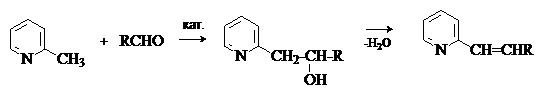Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь FAQ Написать работу КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Билет 3. Вопрос 1. Реакция ВиттигаСодержание книги
Поиск на нашем сайте Билет 3. Вопрос 1. Реакция Виттига Реакция Виттига — химическая реакция альдегидов или кетонов с илидами фосфора (которые иногда называют «реагентами Виттига»), которая приводит к образованию алкенов или алленов и оксида трифенилфосфина[1][2].
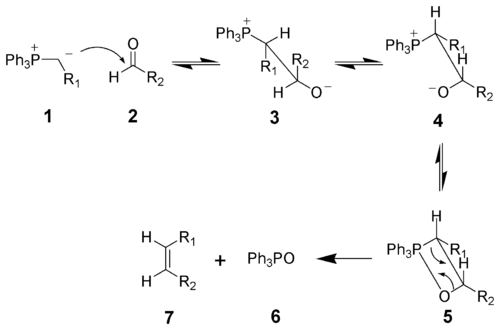
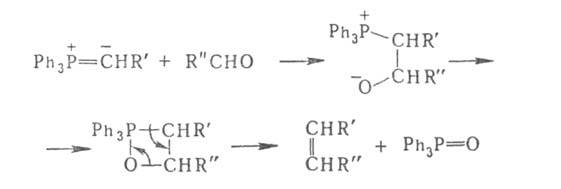
Реакция была открыта в 1954 году Георгом Виттигом. За открытие этой реакции он был награждён Нобелевской премией в области химии в 1979 году. Реакции Виттига широко используется в органическом синтезе для получения алкенов. Классический механизм При нуклеофильном присоединении илида 1 к карбонильному соединению образуется бетаин 3, который вследствие свободного вращения вокруг связи С-С может переходить в конформер 4. Последний способен быстро и обратимо изомеризоваться, образуя четырехчленный оксафосфетановый цикл (соединение 5). Элиминирование оксида трифенилфосфина 6 приводит к образованию Z-изомера целевого алкена 7.
Изомеризация бетаина 4 в оксафосфетан 5 является лимитирующей стадией реакции. Скорость реакции нуклеофильного присоединения илида к карбонильному соединению сильно зависит от природы илида. В случае незамещенного илида (R1 = H) присоединение проходит относительно быстро с подавляющим большинством альдегидов и кетонов. Однако в случае «стабилизированных реагентов Виттига» (R1 = электронноакцепторная группа) скорость нуклеофильного присоединения значительно снижается, что приводит к уменьшению скорости реакции в целом. Также возрастает количество побочного продукта в виде E-изомера алкена. Кроме этого, «стабилизированные реагенты Виттига» практически не взаимодействуют со стерически затрудненными альдегидами и кетонами. ВИТТИГАРЕАКЦИЯ, получение олефинов действием илидов Р (алкилиденфосфоранов) на альдегиды или кетоны.Илиды обычно используют в свежеприготовленном виде. Получают их взаимод. трифенилалкилфосфония с литийорг. соед. или гексаалкилтриамидоалкилфосфония со щелочами. Механизм Виттига реакции можно представить след. образом: Обычно образуется смесь изомерных олефинов. Однако при соответствующем подборе реагентов и условий р-ции (напр., при использовании полярных апротонных р-рителей) возможен синтез преим. одного из изомеров (цис- или транс-изомера). Виттига реакцию широко используют в тонком орг. синтезе, напр. для получения альдегидов, содержащих на один атомС больше, чем в исходном соед., а также полиенов, гетероциклич. соед.: Р-ция открыта Г. Виттигом в 1954.
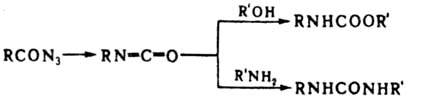
Билет 5. Вопрос 1. Перегруппировка Курциуса КУРЦИУСАРЕАКЦИЯ, получение первичных аминов термич. перегруппировкой ацилазидов в изоцианаты(перегруппировка Курциуса) с послед. их гидролизом: Билет 5. Вопрос 1. Реакция Пааль-Кнорра. ПААЛЯ -КНОРРА РЕАКЦИЯ (р-ция Кнорра, р-ция Кнорра - Пааля), конденсация 1,4-дикарбонильных соед. с NH3 или первичными аминами с образованием пир-ролов:
В р-цию вступают дикетоны, диальдегиды, дикетокарбо-новые к-ты и их эфиры, дикетонитрилы, три- и тетра-кетоны. Стерически затрудненные дикетоны (напр., 4,5-ди-бензоил-1-циклогексен и его замещенные) в р-цию не вступают. Р-цию с низшими алифатич. аминами обычно проводят при комнатной т-ре (р-ритель-Н2О, ROH, RCOOH, С6Н6); с высшими алифатич. и ароматич. аминами процесс осуществляют при нагр. в течение неск. часов, в последнем случае-и в присут. НСl или ледяной СН3СООН. Вместо NH3 и первичных аминов можно использовать аммониевые соли [напр., (NH4)2CO3], мочевину, дициандиамид. В ряде случаев дикетонывовлекают в р-цию в момент выделения, напр.:
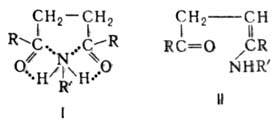
Выходы пирролов 70-90%. В качестве побочных продуктов могут образовываться моно- или диоксимы,олигомеры и полимеры. Механизм Пааля - Кнорра реакции детально не исследован. Полагают, что промежуточно образуются соед. I или II (в нек-рых случаях соед. типа II выделены), к-рые в результате отщепления Н2О образуют пиррол.
При взаимод. с 1,4-дикарбонильными соед. дегидратиру-ющих агентов или P2S5 образуются пятичленные гетеро-циклы (соотв. фураны и тиофены). Эти синтезы иногда также наз. Пааля - Кнорра реакцией:
Пааля - Кнорра реакцию используют в препаративной практике. Синтез пирролов открыт в 1884 Л. Кнорром, впоследствии эта р-ция более подробно исследована К. Паалем.
Билет 7. Вопрос 1. Реакция Вюрца-Фиттига ВЮРЦАРЕАКЦИЯ, конденсация алкилгалогенидов под действием Na (реже - Li или К) с образованием предельных углеводородов: 2RHal + 2Na -> R—R + 2NaHal, где Hal - обычно Br или I. При использовании в р-ции разл. алкилгалогенидов (RHal и R'Hal) образуется трудноразделяемая смесь всех возможных продуктов (R—R, R'—R', R'—R). Вюрца реакция легко протекает, если алкилгалогенид имеет большую мол. массу, а галоген связан с первичным атомомС. Процесс проводят при низких т-рах в сольватирующих р-рителях. Так, в ТГФ р-ция осуществляется быстро и с хорошим выходом уже при — 80 °С. Предполагается, что механизм р-ции включает образование ион-радикалов и радикалов: Однако факт обращения конфигурации нек-рых оптически активных алкилгалогенидов (напр., 2-хлороктана в р-ции с Na) не исключает возможности гетеролитич. механизма. Р-ция открыта Ш. Вюрцем в 1855 и используется гл. обр. для получения углеводородов с длинной углеродной цепью. В др. случаях, особенно при получении несимметричных ал-канов, применяют разл. модификации Вюрца реакции, рассмотренные ниже. Для синтеза жирноароматич. соед. используют модификацию Фиттига (р-цию Вюрца-Фиттига): ArHal + RHal + 2Na -> Ar—R + 2NaHal Р-ция открыта Р. Фиттигом в 1855. Часто с хорошим выходом образуются алканы с использованием реактива Гриньяра, напр.: Несимметричные предельные углеводороды получают, используя медьорг. соед.: Р-цию, подобную Вюрца реакции, используют для синтеза элементоорг. соед. и бициклич. соед., напр.:
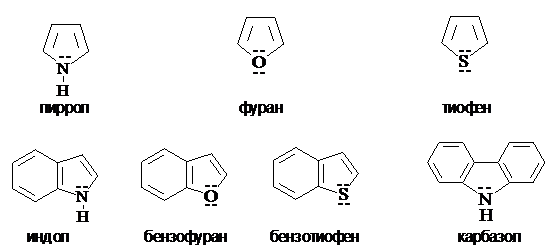
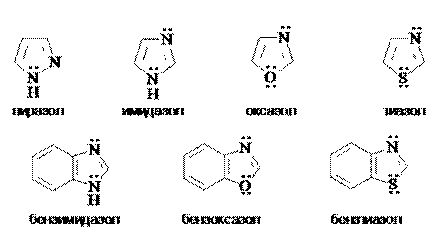
Классификация. Гетероциклы классифицируют по следующим основным признакам: - по природе и числу гетероатомов; - по размеру цикла; - по степени ненасыщенности. Наибольшее распространение имеют пяти- и шестичленные гетероциклы, содержащие в качестве гетероатомов азот, кислород и серу. По степени ненасыщенности различают насыщенные и ненасыщенные, в том числе ароматические гетероциклы. Гетероциклынеароматического характера по своим свойствам сходны с соответствующими ациклическими соединениями (аминами, амидами, простыми и сложными эфирами и т.д.). Ароматические гетероциклические соединения по свойствам близки к бензолу. Для них, как и для бензоидных систем, наиболее характерны реакции замещения. При этом гетероатом выполняет роль «внутренней функции», определяющей скорость и направление реакций замещения. Систематическая номенклатура гетероциклов сложна. Для многих гетероциклических соединений сохраняются тривиальные названия. Ниже приведены некоторые группы ароматических гетероциклических соединений и их тривиальные названия. Пятичленные ароматические гетероциклы с одним гетероатомом
Пятичленные ароматические гетероциклы с двумя гетероатомами
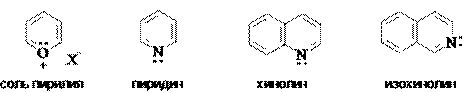
Шестичленные ароматические гетероциклы с одним гетероатомом
Шестичленые ароматические гетероциклы с двумя гетероатомами
Далее будут рассмотрены пяти- и шестичленные ароматические гетероциклы с одним гетероатомом. Методы получения. 1) Синтез из 1,4-дикарбонильных соединений по Паалю-Кнорру.
При нагревании 1,4—дикарбонильных соединений с такими нуклеофильными реагентами, как аммиак или сульфиды, образуются соответственно производные пиррола и тиофена. Нагревание 1,4—дикарбонильных соединений в присутствии кислотного катализатора дает производные фурана. 2) Синтез пирролов по Кнорру.
Метод состоит в конденсации a-аминокетонов с b-дикарбонильными соединениями в присутствии кислотного или основного катализатора. 3) Взаимные превращения (реакция Юрьева). При нагревании (~4000С) над катализатором Al2O3 происходят превращения фурана, пиррола и тиофена друг в друга. Практическое значение имеют получение пиррола и тиофена из фурана.
Химические свойства. 1)Основные свойства Пиррол является чрезвычайно слабым основанием. Протонирование по атому азота означает нарушение стабильной p-системы и потерю ароматичности. В действительности присоединение протона происходит по атомам углерода пиррольного кольца, преимущественно по a-положению. Образующаяся таким образом сопряженная кислота атакует молекулу пиррола, в результате чего происходит полимеризация.
Фуран также полимеризуется под действием кислот. Поэтому пиррол и фуран называют ацидофобными соединениями. Введение электроноакцепторных групп снижает ацидофобность. Например, фурфурол и пирослизевая кислота устойчивы к действию кислот. Тиофен, в отличие от пиррола и фурана, неацидофобен. 2)Кислотные свойства пиррола Пиррол является слабой NH-кислотой (рКа=17,5). По кислотности он превосходит амины и близок к спиртам. Соли пиррола получают взаимодействием пиррола с металлами, амидами металлов, металлоорганическим соединениями.
3)Реакции электрофильного замещения Пиррол, фуран и тиофен являются электронодонорными соединениями и проявляют высокую реакционную способность по отношению к электрофилам. Реакционная способность в реакциях электрофильного замещения возрастает в ряду бензол<тиофен<фуран<пиррол. Электрофильное замещение идет преимущественно в положение 2, что вытекает из сравнения стабильности s-коплексов, ведущих к продуктам 2- и 3-замещения.
Как видно из приведенной выше для пиррола схемы, в s-коплексе (I) достигается более эффективная делокализации положительного заряда (три резонансные структуры), чем в s-коплексе (II) (две резонансные структуры). Взаимодействие гетероциклов с электрофилами может привести и к образованию продуктов присоединения (см. кислотные свойства). Склонность к такого рода превращениям убывает в ряду фуран>пиррол>тиофен, что соответствует степени стабилизации их ароматических систем. Пиррол по реакционной способности по отношению к электрофилам напоминает активированные ароматические субстраты (фенол или ароматические амины). Из-за ацидифобности пиррола при проведении реакций электрофильного замещения необходимо избегатьсильно кислых сред. Основные SE-реакции пиррола суммированы на схеме. Нитрование и сульфирование пиррола возможны только в том случае, если исключена сильнокислая среда. Нитрование проводят действием ацетилнитрата при низких температурах. Для сульфирование используют связанный в комплекс SO3, например,пиридинсульфотриоксид. Галогенирование протекает без катализатора и дает тетрагалогенпирролы.
Пиррол ацилируется ангидридами кислот в отсутствие катализатора. Как активированный субстрат пиррол вступает в реакцииформилирования. Наиболее общим методом формилирования пирролов является реакция Вильсмейера-Хаака.
Фуран, как и пиррол, ацидофобен, поэтому реакции с электрофилами проводят в отсутствие сильных кислот. Фуранацилируетсяангидридами кислот в присутствии мягких кислот Льюиса, сульфируется пиридинсульфотриоксидом, формилируется по реакцииВильсмейера-Хаака. Фуран реагирует более селективно, чем пиррол. Во всех случаях образуются исключительно продукты замещения по положению 2.
Фуран в большей степени, чем пиррол, склонен к образованию с электрофилами продуктов не замещения, а присоединения. Даже реакции, результатом которых является замещение, могут происходить через промежуточное образование продуктов присоединения (по механизму присоединения-отщепления). При нитровании фурана ацетилнитратом можно выделить продукт 2,5-присоединения, который под действием пиридина (Py), подвергается элиминированию с образованием в конечном итоге продукта замещения.
Тиофен, в отличие от пиррола и фурана, неацидофобен, поэтому реакции электрофильного замещения можно проводить с обычнымиэлектрофильными агентами, в том числе с использованием водных растворов минеральных кислот или в присутствии кислот Льюиса. Реакции электрофильного замещения превалируют над процессами присоединения.
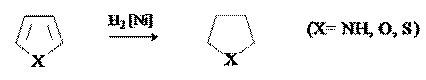
4)Реакции присоединения Гидрирование. Пиррол, фуран и тиофен присоединяют водород в условиях каталитического гидрирования с образованием тетрагидропроизводных.
Труднее других гидрируется тиофен, который отравляет катализатор. Особенностью химии тиофена является восстановительнаядесульфуризация, которая используется в синтетических целях для получения соединений, трудно доступных другими методами.
Реакция Дильса-Альдера Реакции циклоприсоединения характерны для фурана, который в большей степени, чем пиррол и тиофен, проявляет свойства сопряженного диена. Например, он вступает при комнатной температуре в реакцию диенового синтеза с таким активным диенофилом, как малеиновый ангидрид.
Индол. Индол содержит конденсированные бензольный и пиррольный циклы (является бензологом пиррола). Ниже приведены нумерация циклов и примеры систематической и тривиальной номенклатуры производных индола.
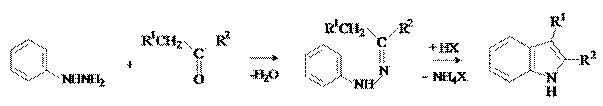
Методы получения. Синтез индолов по Фишеру. Метод состоит в циклизации фенилгидразонов в присутствии кислотного катализатора (H2SO4, ZnCl2). Механизм реакции:
В присутствии кислот происходит изомеризация гидразонов в гидразины с последующей сигматропной перегруппировкой и замыканием пиррольного цикла. Ключевой стадией процесса является разрыв слабой связи N-N и образование прочной связи С-С. Это согласованный процесс, аналогичный перегруппировке Кляйзена аллилфениловых эфиров. Химические свойства. 1)Основные свойства
Индол является очень слабым основанием. Протонирование происходит по положению 3, далее протекает олигомеризация. Таким образом, индол, как и пиррол, является ацидофобным соединением. 2)Реакции электрофильного замещения Все реакции электрофильного замещения в индоле идут по пиррольному кольцу. Существенным отличием от пиррола является ориентация электрофильного замещения в положение 3, что обусловлено более эффективной стабилизацией промежуточнообразующегося катиона.
Катион (I), образующийся при атаке электрофилом по положению 3, эффективно стабилизирован с участием атома азота, тогда как для изомерного катиона (II) невозможна стабилизация без нарушения ароматичности бензольного кольца.
Из-за ацидофобности индола выбор условий электрофильного замещения требует тех же самых предосторожностей, что и в случае пиррола. Кислотные свойства Индол представляет собой слабую NH-кислоту (рКа=17) и образует соли при действии сильных оснований, например:
Методы получения. Синтез хинолина по Скраупу. Метод состоит во взаимодействии первичных ароматических аминов с глицерином в присутствии дегидратирующего реагента и окислителя. Классический синтез Скраупа может быть выражен следующей схемой.
На первой стадии происходит катализируемая серной кислотой дегидратация глицерина с образованием a,b-непредельного альдегида, акролеина. Второй стадия - сопряженное присоединение анилина к акролеину. Третья стадия – замыкание цикла путемэлектрофильной атаки карбонильной группы по ароматическому кольцу. На заключительных стадиях происходит дегидратация и окисление образующегося 1,2-дигидрохинолина в хинолин. В качестве окислителя удобно использовать нитросоединение,соответствующее исходному амину. Для снижения экзотермичности реакции, которая протекает очень бурно, в реакционную смесь добавляют FeSO4. Синтез хинолина по Дебнеру-Миллеру.
Этот метод, как и реакция Скраупа, основан на взаимодействии ароматических аминов с a,b-непредельными карбонильными соединениями. Ненасыщенные карбонильные соединения получают непосредственно в реакционной смеси из альдегидов и кетонов путем кротоновой конденсации. Окислителями промежуточно образующихся 1,2-дигидрохинолинов являются альдимины, образующиеся при взаимодействии исходных карбонильных соединения с ароматическим амином. Химические свойства. 1)Реакции по атому азота. Наличие у атома азота пиридина и его бензологов свободной пары электронов, не участвующей в сопряженной системе, обусловливает их основные и нуклеофильные свойства.
Пиридин и хинолин являются слабыми основаниями и с сильными кислотами образуют соли пиридиния и хинолиния. По основности пиридин и его бензологи уступают алифатическим и алициклическим аминам и близки к ароматическим аминам.
p Для пиридина и его бензологов характерно образование комплексов с кислотами Льюиса, которые в большинстве своем служат мягкими электрофильными агентами, например:
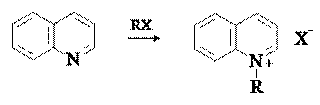
Нуклеофильность атома азота проявляется в его способности к алкилированию. При алкилировании образуются устойчивые солиалкилпиридиния и алкилхинолиния:
Восстановление Каталитическое гидрирование пиридина дает пиперидин. При гидрирование хинолина в первую очередь восстанавливается пиридиниевый цикл и образуется 1,2,3,4-тетрагидрохинолин.
Окисление Ядро пиридина устойчиво к действию окислителей. Алкилпиридины могут быть окислены до пиридинкарбоновых килот. При окислении хинолина и изохинолина в первую очередь, как правило, разрушается бензольное кольцо.
Окисление пиридина и хинолина пероксидом водорода и надкислотами приводит к N-оксидам.
Реакции метилпиридинов Атом азота в пиридиниевом кольце оказывает влияние на подвижность a-водородных атомов в алкилпиридинах. Метилпиридиныобладают большей СН-кислотностью, чем толуол. СН-кислотность атомов водорода метильных групп в 2- и 4-метилпиридинахсравнима с кислотностью метилкетонов. Повышенная протонная подвижность водорода метильных групп в 2- и 4- метилпиридинахможет быть объяснена стабилизацией сопряженных оснований с участием атома азота, что следует из рассмотрения их резонансных структур.
В этом отношении анионы метилпиридинов пободны енолят-анионам. В присутствии оснований 2- и 4- метилпиридины выступают в реакции альдольно-кротоновой конденсации в роли метиленовые компоненты.
Билет 3. Вопрос 1. Реакция Виттига Реакция Виттига — химическая реакция альдегидов или кетонов с илидами фосфора (которые иногда называют «реагентами Виттига»), которая приводит к образованию алкенов или алленов и оксида трифенилфосфина[1][2].
Реакция была открыта в 1954 году Георгом Виттигом. За открытие этой реакции он был награждён Нобелевской премией в области химии в 1979 году. Реакции Виттига широко используется в органическом синтезе для получения алкенов. Классический механизм При нуклеофильном присоединении илида 1 к карбонильному соединению образуется бетаин 3, который вследствие свободного вращения вокруг связи С-С может переходить в конформер 4. Последний способен быстро и обратимо изомеризоваться, образуя четырехчленный оксафосфетановый цикл (соединение 5). Элиминирование оксида трифенилфосфина 6 приводит к образованию Z-изомера целевого алкена 7.
Изомеризация бетаина 4 в оксафосфетан 5 является лимитирующей стадией реакции. Скорость реакции нуклеофильного присоединения илида к карбонильному соединению сильно зависит от природы илида. В случае незамещенного илида (R1 = H) присоединение проходит относительно быстро с подавляющим большинством альдегидов и кетонов. Однако в случае «стабилизированных реагентов Виттига» (R1 = электронноакцепторная группа) скорость нуклеофильного присоединения значительно снижается, что приводит к уменьшению скорости реакции в целом. Также возрастает количество побочного продукта в виде E-изомера алкена. Кроме этого, «стабилизированные реагенты Виттига» практически не взаимодействуют со стерически затрудненными альдегидами и кетонами. ВИТТИГАРЕАКЦИЯ, получение олефинов действием илидов Р (алкилиденфосфоранов) на альдегиды или кетоны.Илиды обычно используют в свежеприготовленном виде. Получают их взаимод. трифенилалкилфосфония с литийорг. соед. или гексаалкилтриамидоалкилфосфония со щелочами. Механизм Виттига реакции можно представить след. образом: Обычно образуется смесь изомерных олефинов. Однако при соответствующем подборе реагентов и условий р-ции (напр., при использовании полярных апротонных р-рителей) возможен синтез преим. одного из изомеров (цис- или транс-изомера). Виттига реакцию широко используют в тонком орг. синтезе, напр. для получения альдегидов, содержащих на один атомС больше, чем в исходном соед., а также полиенов, гетероциклич. соед.: Р-ция открыта Г. Виттигом в 1954.
Билет 5. Вопрос 1. Перегруппировка Курциуса КУРЦИУСАРЕАКЦИЯ, получение первичных аминов термич. перегруппировкой ацилазидов в изоцианаты(перегруппировка Курциуса) с послед. их гидролизом: Билет 5. Вопрос 1. Реакция Пааль-Кнорра. ПААЛЯ -КНОРРА РЕАКЦИЯ (р-ция Кнорра, р-ция Кнорра - Пааля), конденсация 1,4-дикарбонильных соед. с NH3 или первичными аминами с образованием пир-ролов:
В р-цию вступают дикетоны, диальдегиды, дикетокарбо-новые к-ты и их эфиры, дикетонитрилы, три- и тетра-кетоны. Стерически затрудненные дикетоны (напр., 4,5-ди-бензоил-1-циклогексен и его замещенные) в р-цию не вступают. Р-цию с низшими алифатич. аминами обычно проводят при комнатной т-ре (р-ритель-Н2О, ROH, RCOOH, С6Н6); с высшими алифатич. и ароматич. аминами процесс осуществляют при нагр. в течение неск. часов, в последнем случае-и в присут. НСl или ледяной СН3СООН. Вместо NH3 и первичных аминов можно использовать аммониевые соли [напр., (NH4)2CO3], мочевину, дициандиамид. В ряде случаев дикетонывовлекают в р-цию в момент выделения, напр.:
Выходы пирролов 70-90%. В качестве побочных продуктов могут образовываться моно- или диоксимы,олигомеры и полимеры. Механизм Пааля - Кнорра реакции детально не исследован. Полагают, что промежуточно образуются соед. I или II (в нек-рых случаях соед. типа II выделены), к-рые в результате отщепления Н2О образуют пиррол.
При взаимод. с 1,4-дикарбонильными соед. дегидратиру-ющих агентов или P2S5 образуются пятичленные гетеро-циклы (соотв. фураны и тиофены). Эти синтезы иногда также наз. Пааля - Кнорра реакцией:
Пааля - Кнорра реакцию используют в препаративной практике. Синтез пирролов открыт в 1884 Л. Кнорром, впоследствии эта р-ция более подробно исследована К. Паалем.
Билет 7. Вопрос 1. Реакция Вюрца-Фиттига ВЮРЦАРЕАКЦИЯ, конденсация алкилгалогенидов под действием Na (реже - Li или К) с образованием предельных углеводородов: 2RHal + 2Na -> R—R + 2NaHal, где Hal - обычно Br или I. При использовании в р-ции разл. алкилгалогенидов (RHal и R'Hal) образуется трудноразделяемая смесь всех возможных продуктов (R—R, R'—R', R'—R). Вюрца реакция легко протекает, если алкилгалогенид имеет большую мол. массу, а галоген связан с первичным атомомС. Процесс проводят при низких т-рах в сольватирующих р-рителях. Так, в ТГФ р-ция осуществляется быстро и с хорошим выходом уже при — 80 °С. Предполагается, что механизм р-ции включает образование ион-радикалов и радикалов: Однако факт обращения конфигурации нек-рых оптически активных алкилгалогенидов (напр., 2-хлороктана в р-ции с Na) не исключает возможности гетеролитич. механизма. Р-ция открыта Ш. Вюрцем в 1855 и используется гл. обр. для получения углеводородов с длинной углеродной цепью. В др. случаях, особенно при получении несимметричных ал-канов, применяют разл. модификации Вюрца реакции, рассмотренные ниже. Для синтеза жирноароматич. соед. используют модификацию Фиттига (р-цию Вюрца-Фиттига): ArHal + RHal + 2Na -> Ar—R + 2NaHal Р-ция открыта Р. Фиттигом в 1855. Часто с хорошим выходом образуются алканы с использованием реактива Гриньяра, напр.: Несимметричные предельные углеводороды получают, используя медьорг. соед.: Р-цию, подобную Вюрца реакции, используют для синтеза элементоорг. соед. и бициклич. соед., напр.:
|
||||||||||
|
|
Последнее изменение этой страницы: 2016-12-10; просмотров: 479; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 216.73.216.108 (0.013 с.) |











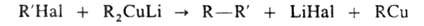























 11,25 5,23 5,14 4,94 4,58
11,25 5,23 5,14 4,94 4,58