Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Химические канцерогенные факторы.
Наиболее распространенной классификацией химических канцерогенных веществ в настоящее время является разделение их на классы в соответствии с химическим строением: 1) полициклические ароматические углеводороды (ПАУ) и гетероциклические соединения; 2) ароматические азосоединения; 3) ароматические аминосоединения; 4) нитрозосоединения и нитрамины; 5) металлы, металлоиды и неорганические соли. Канцерогенными свойствами могут обладать и другие химические вещества. Опухолевая трансформация начинается не сразу после контакта канцерогена с клеткой: вначале канцерогенное вещество подвергается биотрансформации, в результате образуются канцерогенные метаболиты, которые внедряются в клетку, изменяют ее генетический аппарат, обусловливая малигнизацию. Биологические канцерогенные факторы. Наиболее изученными среди биологических канцерогенов являются вирусы. К началу 60-х гг. ХХ в. Л.А. Зильбер окончательно сформулировал вирусогенетическую концепцию развития опухолевого процесса, согласно которой онкогенный вирус, попадая в клетку, внедряет свой генетический материал в состав хромосомы клетки-хозяина, становясь ее интегральной частью («геном» или «батареей генов») и тем самым индуцируя трансформацию нормальной клетки в опухолевую. В зависимости от химической природы генетического материала онкогенные вирусы подразделяются на ДНК- и РНК-содержащие. Среди ДНК-содержащих онкогенных вирусов выделяют следующие группы. Паповавирусы. К ним относятся папилломатозные вирусы, которые вызывают доброкачественные новообразования (папилломы) кожи и слизистой оболочки у различных видов животных (мыши, кролики, свиньи, кошки, собаки) и человека. Аденовирусы. Герпес-вирусы вызывают злокачественные опухоли у животных и человека: вирус Эпштейна-Барр определяет возникновение лимфомы Беркитта и рака носоглотки; вирус гепатита С вызывает рак печени. Вирусы группы оспы РНК-содержащие онкогенные вирусы (ретровирусы, онкорнавирусы). В отличие от ДНК-содержащих вирусов они являются естественными возбудителями большинства злокачественных опухолей у животных. Канцерогенез, индуцируемый ДНК- и РНК-содержащими вирусами, включает следующие основные этапы: 1) вирус проникает в клетку и закрепляет свой генетический материал в клетке-хозяине путем физической интеграции с клеточной ДНК; 2) начинается экспрессия специфических генов (онкогенов) в составе интегрированного вирусного генома с образованием специфических матричных РНК (мРНК) и онкобелков, которые ответственны за превращение нормальной клетки в опухолевую; 3) под влиянием онкобелков клетка утрачивает чувствительность к факторам, регулирующим деление, и по своим фенотипическим признакам (морфологическим, цитогенетическим, биохимическим) становится опухолевой.
К биологическим особенностям опухолевых клеток и тканей (в первую очередь - злокачественных) относятся беспредельность роста, автономность роста, инфильтрирующий рост, способность к метастазированию, атипизм (анаплазия) опухолевых клеток и тканей, клоновый характер роста, опухолевая прогрессия. 1)Беспредельность роста - это избыточность пролиферации опухолевых клеток. Это обусловливается тем, что в опухоли контактное торможение заблокировано (в норме беспредельному размножению препятствует контактное торможение соседних клеток); в опухолевых клетках антионкогены инактивированы, поэтому программа апоптоза не включается; в опухолевых клетках активируются протоонкогены, которые кодируют ростовые факторы. 2)Автономность роста. Обусловлена снижением на опухолевых клетках числа рецепторов к гормонам и нейромедиаторам организма и переходом к пара- и аутокринной регуляции пролиферации. 3)Инфильтрирующий (инвазивный) рост - это критерий злокачественности опухолевого роста. При инфильтрирующем росте опухолевые клетки выходят за пределы исходной ткани, прорастают в окружающие ткани, разрушая их при этом. Для доброкачественных опухолей характерен экспансивный рост. 4)Метастазирование - также один из критериев злокачественности опухоли. Это появление вторичных новых очагов опухолевого роста, удаленных от первичного опухолевого узла. 5)Атипизм (анаплазия) - это особенности структурно-функциональной организации опухолевых клеток, создающие сходство с эмбриональными клетками и отличающие их от нормальных исходных клеток.
Морфологический атипизм. Для злокачественных опухолей характерен как клеточный, так и тканевой атипизм. Первый заключается в необычной величине, форме и строении опухолевых клеток. При их разрастании отмечаются омоложение клеток, возврат их структуры к наиболее примитивной, эмбриональной организации. Ядра этих клеток огромные, уродливой формы, с изрезанными границами и неравномерно распределенным по нуклеоплазме хроматином, в них чаще происходят митозы. Метаболический атипизм. Опухоли потребляют меньше кислорода, чем нормальные ткани. В опухоли постоянно обнаруживается 10-30-кратное усиление анаэробного гликолиза. Усиление гликолиза и ослабление тканевого дыхания прогрессивно нарастают по мере увеличения степени злокачественности опухоли. В опухоли проявляется преобладанием липогенеза над липолизом, при этом особенно интенсивно синтезируются липиды и липопротеины, которые в дальнейшем идут на построение мембран вновь образующихся клеток. Для опухоли характерно преобладание анаболизма над катаболизмом белков, что приводит к возрастанию уровня протеинов, необходимых для усиленного размножения клеток. Функциональная анаплазия - это особенности функционирования опухолевых клеток по сравнению с исходными нормальными клетками. Возможны следующие ее варианты: 1. Снижение либо полная утрата специализированной функции, свойственной нормальным клеткам и тканям. 2. Сохранение функции. Но она выполняется монотонно, некоординированно с организмом, не соответствует его потребностям 3. Появление новой несвойственной функции. Иммунологическая анаплазия – изменение антигенных свойств опухолевых клеток. Существует 3 варианта изменения набора антигенов опухолевых клеток: 1) антигенное упрощение - уменьшение количества антигенов; 2) антигенная дивергенция - появление у опухолевых клеток антигенов различных нормальных клеток и тканей; 3) антигенная реверсия - появление эмбриональных антигенов. Все это становится одним из механизмов ускользания опухоли от иммунного надзора организма. 6)Клональное развитие. Большинство известных опухолей развиваются из одной опухолевой клетки, возникшей вследствие ее соматической мутации, и характеризуются моноклональным происхождением. Опухоли поликлонального происхождения характеризуются ростом из нескольких клеток и образованием нескольких зачатков опухолей. 55. Основные теории генеза опухолевого роста (см. вопрос 54 Физические, химические, биологические канцерогенные факторы, по сути то же самое). Протоонкогены - специфические гены нормальных клеток, которые осуществляют позитивный контроль процессов пролиферации и мембранного транспорта. Под влиянием мутаций протоонкогены претерпевают так называемую активацию, что способствует их превращению в онкогены, экспрессия которых вызывает возникновение и прогрессию опухолей. Протоонкогены кодируют белки, которые запускают цепь последовательных сигналов для нормального роста и функционирования клеток. В результате мутационного превращения протоонкогенов в онкогены одно или несколько звеньев этой цепи спонтанно становятся сверхактивными. Онкоген — это ген, кодирующий белок, который, в случае нарушения регуляции, может вызвать образование злокачественной опухоли. Мутации, вызывающие активацию онкогенов, повышают шанс того, что клетка превратится в раковую клетку. Продукты деятельности онкогенов — онкобелки. Многие онкобелки гомологичны или родственны ростовым факторам.
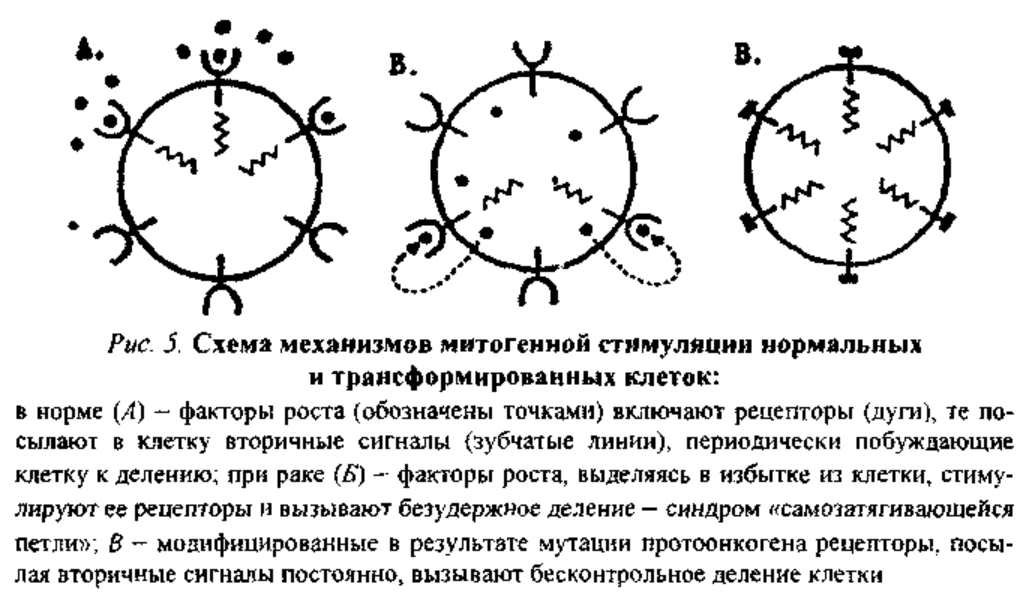
Механизмы действия онкогенов и их продуктов — онкобелков можно подразделить на три основные категории (рис. 5). Антибластомной резистентностью называется устойчивость организма к опухолевому росту. Различают три группы механизмов антибластомной резистентности. 1. Антиканцерогенные механизмы, действующие на этапе взаимодействия канцерогенного агента с клетками: инактивация химических канцерогенов в микросомальной системе; их элиминации из организма в составе желчи, мочи, кала; выработка антител к соответствующим канцерогенам; ингибирование свободнорадикальных процессов и перекисного окисления липидов (антирадикальные и антиперекисные реакции), обеспечиваемое витамином Е, селеном, супероксиддисмутазой и др.; взаимодействие с онкогенными вирусами интерферона, антител и др. 2. Антимутационные механизмы: поддержание генного гомеостаза за счет процессов репарации ДНК; синтез ингибиторов опухолевого роста, обеспечивающих подавление размножения клеток и стимуляцию их дифференцировки (функция антионкогенов). 3. Антицеллюлярные механизмы, направленные на ингибирование и уничтожение отдельных опухолевых клеток, на предотвращение образования их колонии, т.е. опухоли. К ним относятся иммуногенные механизмы – неспецифические (реакция ЕК) и специфические (реакция иммунных Т-киллеров; иммунных макрофагов), неимунногенные факторы и механизмы (ФНО, ИЛ-1, торможения аллогенное, контактное, кейлонное - регулирующее нейротрофическое и гормональное влияние - и др.).
56. Голодание – это состояние, возникающее в тех случаях, когда организм не получает пищевых веществ совсем, или получает их в недостаточном количестве, или же не усваивает их вследствие болезни. Выделяют следующие виды голодания: 1. Абсолютное голодание – полное прекращение поступления в организм пищи и воды; 2. Полное голодание – полное отсутствие приема пищи при сохранении приема воды; 3. Неполное голодание – состояние, характеризующееся тем, что калорийность принимаемой пищи не покрывает всех энергетических затрат организма; 4. Частичное голодание – калорийность принимаемой пищи полностью покрывает энергетические затраты организма, однако в составе пищи отсутствуют или имеются в недостаточном количестве те или иные питательные вещества (Б, Ж, У, витамины, минеральные соединения и др.).
В процессе полного голодания выделяется три периода: начальный (длится 2-4 дня), стационарный (50-60 суток) и терминальный (2-3 суток). Обмен веществ при голодании. Первый период голодания характеризуется усиленным расходованием углеводов, уровень глю в крови снижается => снижение секреции инсулина => повышение выделения глюкагона. Содержание гликогена в печени быстро снижается, ослабляется эффективность цикла Кребса (т.к. мало инсулина). Выделение азота с мочой уменьшается уже на 2-3-й день голодания. Затем на 5-6-й день происходит переключение обмена на жиры, наблюдается кратковременное повышение выделения азота с мочой, после оно снижается. Снижается биосинтез а.к. из а-кетокислот и аммиака. Однако все эти процессы не могут сбалансировать распад белков => отрицательный азот. баланс. Во втором периоде происходит преимущественное окисление жиров. Мало инсулина => в липоцитах мало глюкозы => недостаток глицерина для синтеза триглицеридов => усиливается липолиз => липемия (жир в крови) => в печени и мышцах повышается уровень свободных жирных кислот, из-за недостатка белков развивается недостаток липопротеинов => жировая инфильтрация печени. Истощение гликогена =>уменьшение малонин-СоА (подавляет транспорт жира в митохондрии) => повышается продукция кетоновых тел. Происходит глубокая перестройка обменных процессов, направленная на лучшее использование резервных веществ, органические потребности тех органов, которые имеют меньшее значение для сохранения жизни. Третий период характеризуется резким усилением распада белков жизненно важных органов, расходуемых в качестве энергетического материала. Увеличивается выведение с мочой азота, калия, серы, фосфора. Возникают деструктивные изменения в митохондриях. В результате снижения онкотического давления крови (из-за гипопротеинемии) происходит задержка воды (отеки). Из-за тружности восстановления белков (ферментов) происходит расстроиство ферментных систем. Органы и системы при голодании. Со стороны нервной системы в первом периоде отмечается возбуждение, особенно пищевого центра. В дальнейшем развивается угнетение, рефлексы снижаются. В начале голодания повышается функция щитовидной железы, гипофиза, увеличивается секреция кортикотропина и тиротропина, что стимулирует надпочечники. Во втором периоде функции большинства эндокринных желез угнетается. Деятельность пищеварительной системы угнетается.
57. Нарушения основных этапов белкового обмена. Патофизиология нарушений биосинтеза белковых структур. Нарушения биосинтезамогут возникать: 1. В результате недостаточного поступления белков с пищей (при голодании и алиментарной недостаточности) (см. вопрос 56);
2. При патологии процессов расщепления белков в ЖКТ и при нарушении всасывания а.к.; 3. При нарушении синтеза белка в клетках. Патология расщепления и всасывания Причиной патологии процессов белкового синтеза являются заболевания пищеварительного тракта, которые приводят к нарушению расщепления белковых компонентов пищи и всасывания а.к. Такие состояния могут возникать при оперативных вмешательствах на желудке и кишечнике. Например, при удалении значительной части желудка нарушение обмена белков будет связано с тем, что нарушается эвакуационная функция оставшейся части желудка => пищевой комок не подготавливается в достаточной степени к переработке протеолитическими ферментами кишечника. При панкреатитах, камнях и опухолях протока поджелудочной железы возникают нарушения всасывания а.к. При энтеритах нарушается выработка кишечных ферментов => нарушается расщепление белков до а.к. => нарушение всасывания и синтеза белков. Нарушение синтеза белка в клетке Все патологические состояния, приводящие к затруднению мембранного транспорта, неизбежно будут сказываться на синтезе белка, т.к. надо доставить а.к. к рибосомам. К таким патологическим процессам относятся некоторые эндокринные расстройства. При сахарном диабете будет наблюдаться торможение белкового синтеза, т.к. инсулин обеспечивает транспорт а.к. в клетку. СТГ способствует образованию комплексов рибосом, на которых идет сборка белка => при гипофункции передней доли гипофиза ведет к снижению кол-ва СТГ и торможению биосинтеза белка. Глюкокортикоиды, которые вырабатываются корой надпочечников, во всех органах, кроме печени, на белковый синтез оказывают антианаболическое действие, т.е. они его тормозят. В печени они усиливают синтез белка => патология коры надпочечников, ведущая к ее гипофункции, сказывается на синтезе белков в печени. Со стороны нервной системы: При денервации органов в них резко снижается интенсивность белкового синтеза, что в дальнейшем приводит к развитию трофических расстройств – появлению в паренхиме органа атрофических изменений, образованию язв и т.д. Азотистый баланс – параметр, характеризующий соотношение анаболизма и катаболизма белка в организме. Положительный азотистый баланс свидетельствует о том, что поступление с пищей азота превышает его выведение из организма (в период роста, при беременности, в период выздоровления после болезни). При недостаточном поступлении калорий с пищей (при голодании) белки тканей разрушаются, обеспечивая энергетические потребности и глюконеогенез => в организм поступает меньше азота, чем выводится. Такое состояние называют отрицательным азотистым балансом (при изнурительных заболеваниях, когад белки пищи неполноценны и т.д.).
58. Нарушения обмена аминокислот. Нарушение трансаминирования и дезаминирования. Нарушения реакции трансаминирования могут возникать из-за недостаточности пиридоксина (кофактора ферментов аминотрансферазы и трансаминазы). Гликокортикоиды и гормон щитовидной железы оказывают стимулирующее действие на трансаминирование => при их недостатке изменяется скорость трансаминирования. Угнетение дезаминирования приводит к накоплению а.к. => гипераминоацидемия => аминоацидурия => изменение соотношения а.к. => неблагоприятные условия для синтеза белка. Нарушение дезаминирования может происходить из-за недостатка компонентов, которые участвуют в этом процессе (пиридоксин, рибофлавин и др.).
Наследственные нарушения обмена некоторых а.к. Нарушения обмена фенилаланина. В норме фенилаланин окисляется в тирозин. Если же в печени нарушается синтез необходимого для этого фермента фенилаланингидроксилазы, то окисление фенилаланина идет по пути образования фенилпировиноградной и фенилмолочной кислот – развивается фенилкетонурия. Также фенилаланин накапливается в крови, тканях, цереброспинальной жидкости, что ведет к тяжелому поражению ЦНС. Нарушение обмена тирозина. В норме тирозин превращается в парагидроксифенилпировиноградную кислоту, а затем в гомогентизированную кислоту, затем (под действием оксидазы гомогентизированной кислоты, которая синтезируется в печени) в малеилацетоуксусную кислоту. При недостатке превращения парагидроксифенилпировиноградной кислоты в гомогентизированную кислоту, они выделяются с мочой => тирозиноз. При задержке превращения гомогентизированной кислоты в метилацетоуксусную кислоту, развивается алкаптонурия. Моча при стоянии на воздухе и при добавлении к ней щелочи становится черной, из-за окисления гомогентизиновой кислоты. Тирозин также превращается в меланин. Если в организме недостаток тирозиназы => мало меланина => альбинизм. Подагра (болезнь королей, пиратская болезнь, болезнь воскресного вечера и утром в понедельник) – хроническое метаболическое заболевание, связанное с нарушением пуринового обмена и накоплением в организме мочевой кислоты, клинически проявляющееся рецидивирующим артритом, образованием подагрических узлов (тофусов) и поражением внутренних органов. По этиологии различают подагру первичную и вторичную. Первичная подагра является наследственным, сцепленным с Х-хромосомой рецессивным заболеванием; болеют в основном мужчины. Генетический дефект обусловливает резкое повышение активности фермента 5-фосфорибозил-1-пирофосфат-синтетазы (ФРПФ-синтетазы) или частичную потерю активности фермента гипоксантин-гуанин-фосфорибозил-трансферазы. В том и другом случае происходит избыточное образование и повышенная экскреция уратов. Вторичная подагра обусловлена длительно существующей гиперурикемией приобретенного характера (главный этиологический фактор). Риск развития вторичной подагры увеличивается в регионах с повышенным содержанием молибдена в почве и воде. Комплексными способствующими факторами являются алкоголь (содержит пурины, подкисляет среду, ограничивает экскрецию уратов с мочой), стресс, травмы. Патогенез. При подагре наблюдается нарушение соотношения синтеза и выделения мочевой кислоты из организма. Гиперурикемия приводит к проникновению уратов в синовиальную жидкость и выпадению их в виде кристаллов (проникают в хрящ и синовиальную оболочку, где откладываются в виде игольчатых образований), через дефекты хряща мочевая кислота проникает до субхондральной кости, где образуются тофусы. С другой стороны, экскреция почками уратов снижается. Осаждающиеся соли мочевой кислоты активируют систему комплемента и фактор Хагемана, а через них - кининовую, свертывающую и фибринолитическую системы. Образующиеся хематтрактанты привлекают лейкоциты, фагоцитирующие кристаллы уратов. Гибель нейтрофилов в процессе фагоцитоза сопровождается выделением в окружающие ткани лизосомальных ферментов и активных радикалов кислорода. Макрофаги, также принимающие активное участие в фагоцитозе, выделяют провоспалительные цитокины и другие медиаторы. Как следствие этих явлений возникает острое воспаление (острый подагрический артрит), в дальнейшем приобретающее хронический характер.
59. Нарушения углеводного обмена. При нарушении углеводного обмена могут развиваться состояния гипергликемии (концентрация глюкозы в крови более 5,5 ммоль/л) и гипогликемии (менее 3,3 ммоль/л). Патогенез гипогликемий может быть связан с недостаточным поступлением глюкозы в кровь, ускоренным выведением ее из крови либо комбинацией этих факторов. Различают физиологическую и патологическую гипогликемию. Физиологическая гипогликемия. Выявляется при тяжелой и длительной физической нагрузке; длительном умственном напряжении; у женщин в период лактации; развивается сразу вслед за алиментарной гипергликемией благодаря компенсаторному выбросу в кровь инсулина. Патологическая гипогликемия (гиперинсулинизм). Чаще возникает у больных сахарным диабетом в связи с передозировкой инсулина при лечении. Причиной ее могут быть также: аденома островковых клеток поджелудочной железы (инсулома); синдром Золлингера- Эллисона (аденома или карцинома поджелудочной железы, которая, по-видимому, развивается из α-клеток островков Лангерганса, ответственных за выделение глюкагона и гастрина). Патологическая гипогликемия (без гиперинсулинизма). Выявляется: при патологии почек, сопровождающейся снижением порога для глюкозы, что приводит к потере глюкозы с мочой; нарушении всасывания углеводов; заболеваниях печени, сопровождающихся торможением синтеза гликогена и глюконеогенеза (острые и хронические гепатиты); недостаточности надпочечников (дефицит глюкокортикоидов); гипоавитаминозе В1, галактоземиях и при печеночных формах гликогенозов; голодании или недостаточном питании (алиментарная гипогликемия); недостаточности механизмов регуляции углеводного обмена у новорожденных. Гипергликемия у человека встречается чаще, чем гипогликемия. Различают следующие типы гипергликемий. Физиологические гипергликемии. Это быстрообратимые состояния. Нормализация уровня глюкозы в крови происходит без каких-либо внешних корригирующих воздействий. К ним относятся: 1. Алиментарная гипергликемия. Обусловлена приемом пищи, содержащей углеводы. Концентрация глюкозы в крови нарастает вследствие ее быстрого всасывания из кишечника. 2. Нейрогенная гипергликемия. Развивается в ответ на эмоциональный стресс и обусловлена выбросом в кровь большого количества катехоламинов, образующихся в мозговом веществе надпочечников и реализующих свои гипергликемические эффекты. Освобождающаяся глюкоза быстро выходит в кровь, обусловливая гипергликемию. Физиологический смысл этого феномена состоит в обеспечении срочной мобилизации резерва углеводов для использования их в качестве источников энергии (окисления) в предстоящей повышенной двигательной активности в условиях стресса. Патологические гипергликемии. Причинами их развития являются: 1) нейроэндокринные расстройства, когда нарушены соотношения уровня гормонов гипо- и гипергликемического действия. 2) органические поражения центральной нервной системы, расстройства мозгового кровообращения различной этиологии; 3) нарушения функций печени при циррозе; 4) судорожные состояния, когда происходит расщепление гликогена мышц и образование лактата, из которого в печени синтезируется глюкоза; 5) действие наркотических веществ (морфин, эфир), возбуждающих симпатическую нервную систему и тем самым способствующих развитию гипергликемии. Сахарный диабет (СД) - это группа метаболических (обменных) заболеваний, характеризующихся гипергликемией, которая является результатом дефектов секреции инсулина, действия (активности) инсулина или обоих этих факторов. Выделяют инсулинзависимый (диабет 1 типа) и инсулиннезависимый (диабет 2 типа). Инсулинзависимый сахарный диабет характеризуется инсулинпенией, т.е. абсолютной инсулиновой недостаточностью, существенными метаболическими нарушениями с тенденцией к кетоацидозу. Инсулиннезависимый сахарный диабет. В его основе лежит относительная инсулиновая недостаточность, провоцируемая перееданием, ожирением, уменьшением числа рецепторов к инсулину. АТ в в-клеткам в поджелудочной железе и инсулину отсутствуют. Этиология. Причиной сахарного диабета является инсулиновая недостаточность. Она мб панкреатической, т.е. связанной с нарушением биосинтеза и выделения инсулина, или внепанкриатической при нормальном выделении инсулина панкреатическими островками. Инсулиновая недостаточность мб связана с генетическими либо приобретенными факторами. Панкреатическая инсулиновая недостаточность возникает в результате нарушений на любой стадии образования и секреции инсулина: повреждение глюкорецепторной системы, когда прекращается выброс инсулина в кровь в ответ на раздражение глюкозой мембраны в-клеток; результат нарушения механизма поступления кальция в клетку, что затрудняет передачу информации с рецептора на клетку; врожденные и приобретенные поломки в соответствующей части ген. аппарата; дефицит необходимых для синтеза инсулина а.к.; нарушение перехода проинсулина в инсулин и секреции инсулина в-гранулами в-клеток. Нарушение целости панкреатических островков при различных деструктивных процессах, инфекционное поражение островков, иммунная реакция на инсулин, либо в-клетки. Внепанкреатическая или относительная инсулиновая недостаточность возникает в тех случаях, когда появляются факторы, угнетающие действие инсулина или ускоряющие его катаболизм. Например, повышенная продукция контринсулярных гормонов (глюкагон). К инактивации инсулина могут привести повышенная активность инсулиназы, хронические воспалительные процессы, АТ к инсулину, отсутствие ферментов, освобождающих инсулин от связи с сывороточными белками. В ряде случаев заболевание обусловлено состоянием инсулиновых рецепторов. При сахарном диабете может снижаться число рецепторов или их сродство к инсулину. Патогенез. Основной механизм: замедление скорости гексокиназной реакци, обусловленное снижением проницаемости клеточных мембран => и транспорта глю в клетки, а также понижением активности гексокиназы в клетках. Это приводит к замедлению образования глюкозо-6-фосфата, а затем и использования этого первого метаболита обмена глю на всех путях превращения его в клетке. Активируется глюконеогенез. Гипергликемия – фактор диабетических ангиопатий. Они проявляются в виде склероза, облитерации и др. поражений кровеносных сосудов. Гипергликемия сопровождается повышением концентрации глико- и мукопротеидов, которые легко выпадают в соединительной ткани, способствуя образованию гиалина и поражению сосудистой стенки. Гипергликемия приводит к гликозурии. Повышение осмотического давления мочи способствует полиурии => вызывает обезвоживание => усиленная жажда. Снижается образование жира из углеводов. Усиливается липолиз и выход жирных кислот из жтровой ткани. Происходит усиленное образование кетоновых тел, т.к. ацетил-СоА не может полностью превратиться в цитрат и сгореть в цикле Кребса. Усиливается катаболизм белков с использованием дезаминированных а.к. для образования углеводов (гликонеогенез). Замедление синтеза белка и ускорение гликонеогенеза способствуют развитию отрицательного азотистого баланса. Патогенез диабетической комы. Диабетическая кома характеризуется потерей сознания, глубоким расстройством обмена веществ и нарушениями рефлекторный деятельности. В патогенезе диабетической комы играет роль целый ряд факторов, из которых на первое место надо поставить кетоз, поскольку накопление кислых кетоновых тел наряду с резким возрастанием в крови концентрации СЖК и молочной кислоты ведет к развитию метаболического ацидоза, рН крови смещается в кислую сторону. Важную роль играет обезвоживание организма, причем, поскольку при недостатке инсулина калий плохо усваивается клетками, возникает гиперосмолярная гипогидрия: осмотическое давление во внеклеточном пространстве нарастает, вода начинает выходить из клеток, которые сморщиваются и гибнут. Дегидратация ведет к сгущению крови, что вызывает серьезные циркуляторные расстройства в системе кровообращения. Одним из важных патогенетических механизмов диабетической комы является энергетическое голодание тканей, в первую очередь, клеток ЦНС.
60. Нарушения основных этапов жирового обмена. Нарушение переваривания и всасывания липидов Причинами нарушения переваривания и всасывания липидов являются: 1. Дефицит или низкая активность панкреатической липазы (поражение поджелудочной железы), что приводит к нарушению расщепления жиров. 2. Недостаточное поступление желчных кислот в кишечник (при гепатитах, циррозах, холециститах, обтурационной желтухе и др.) вызывает нарушение эмульгирования и расщепления жира, а также переноса продуктов его гидролиза к всасывающей поверхности эпителия кишечника. 3. Дефицит гормонов желудочно-кишечного тракта (холецистокинин, гастрин и др.), регулирующих сокращение стенок желчного пузыря, процессы эмульгирования и расщепления жиров, их транспорт через кишечную стенку. 4. Поражение эпителия тонкого кишечника различными ядами и инфекционными агентами, инактивирующими ферментные системы ресинтеза триацилглицеролов эпителия тонкого кишечника, а также процессы фосфорилирования и дефосфорилирования в стенке кишечника. 5. Авитаминозы А, В, С (поскольку эти витамины являются коферментами соответствующих биохимических реакций). 6. Избыточное потребление с пищей Са и Mg, что приводит к образованию нерастворимых в воде солей жирных кислот (мыла). 7. Дефицит холина в пище или недостаточное его образование из метионина при малобелковом питании тормозит реабсорбцию липидов. 8. Изменение деятельности нервной и эндокринной систем: перерезка блуждающего нерва ослабляет всасывание жиров из кишечника, аналогично действует наркоз; адренокортикотропный гормон (АКТГ) и тироксин усиливают всасывание жира. При недостатке гормонов коры надпочечников или избытке адреналина всасывание жира замедляется. 9. Усиленная перистальтика кишечника и диарея препятствуют реабсорбции большей части жира. Организм может терять липиды с мочой (липидурия), что наблюдается при липоидном нефрозе. Нарушение транспорта липидов
|
|||||||||
|
|
Последнее изменение этой страницы: 2021-06-14; просмотров: 69; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.9.141 (0.063 с.) |