Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Этиология и патогенез гемохроматоза
Гемохроматоз – заболевание, характеризующееся избыточным всасыванием железа и его накоплением в клетках паренхиматозных органов с последующим их повреждением. Впервые термин «гемохроматоз» был предложен в XIX в. фон Реклингхаузеном. Частота встречаемости гемохроматоза в европейской популяции –1/300, причем чаще встречается у испанцев и потомков кельтов. Классификация гемохроматоза (Beutler E., 2003): 1. Наследственный (идиопатический, первичный) гемохроматоз ü Тип 1 – классический гемохроматоз ü Тип 2 - ювенильный гемохроматоз ü Тип 3 – гемохроматоз, обусловленный дефицитом рецепторов к трансферрину 2-го типа ü Тип 4 – гемохроматоз, связанный с недостаточным образованием ферропортина-1 ü Тип 5 – африканский гемосидероз (гемосидероз банту) 2. Вторичный гемохроматоз А) встречающийся при наследственных заболеваниях ü талассемии ü дефиците пируваткиназы ü анемиях вследствие нарушения эритропоэза ü дефиците глюкозо-6-фосфат-дегидрогеназы ü наследственном сфероцитозе ü сидеробластных анемиях б) наблюдающийся при приобретенных формах патологии ü сидеробластных и других дисэритропоэтических анемиях ü анемиях, при лечении которых используются частые гемотрансфузии (за исключением острых и хронических постгеморрагических анемий). Классическим наследственным гемохроматозом (тип 1) чаще страдают мужчины. Заболевание чаще выявляется у них после 40 лет. Классический гемохроматоз обусловлен мутацией гена HFE. Этот ген расположен в коротком плече 6-й хромосомы и связан с генами 1-го класса молекул гистосовместимости. Ген HFE экспрессирован в клетках, расположенных в криптах слизистой оболочки тонкого кишечника, а также в макрофагах. Ген HFE кодирует образование соответствующего белка. Этот белок в эндоплазматическом ретикулуме клеток формирует комплекс с b2-микроглобулином. Образовавшийся гетеродимер перемещается к клеточной мембране, при нейтральных значениях рН образуя стабильный комплекс с рецептором для трансферрина и снижая аффинность трансферрина к этому рецептору. Описано несколько видов мутаций гена HFE, наследуемых по аутосомно-рецессивному типу – C282Y, H63D, S65C и др. Чаще всего встречается миссенс-мутация, при которой в 282-м положении аминокислотной последовательности полипептидной цепи цистеиновый остаток заменяется на тирозиновый. В результате изменяется конформация белка HFE и он утрачивает свои свойства, что сопровождается увеличением сродства трансферрина к его рецептору. В кишечнике всасываются большие количества железа, и в виде ферритина и гемосидерина оно депонируется в клетках паренхиматозных органов и макрофагах.
Ювенильный гемохроматоз (тип 2) обусловлен мутацией гена, кодирующего образование белка гепцидина, либо мутацией пока неидентифицированного гена, расположенного в длинном плече 1-й хромосомы. Заболевание начинается в юношеском возрасте и быстро прогрессирует. Железо откладывается преимущественно в миокарде и эндокринных железах. Пациенты не доживают до 4-го десятилетия, так как погибают от сердечной недостаточности. 3-й тип гемохроматоза связан с дефицитом рецепторов к трансферрину 2-го типа. В физиологических условиях эти рецепторы обеспечивают эндоцитоз трансферрина, несущего Fe3+. 4-й тип гемохроматоза обусловлен недостаточным образованием и / или нарушением функций белка ферропортина-1. Этот гликопротеин в физиологических условиях транспортирует железо к базолатеральной мембране энтероцитов, а также удаляет железо из цитоплазмы макрофагов и гепатоцитов. Африканский тип гемохроматоза особеннно широко распространен среди мужчин народности банту. Он возникает в результате мутаций генов, кодирующих образование железо-транспортных белков (чаще – ферропортина). Существенная роль в его патогенезе принадлежит также увеличению поступления железа с пищей. Это происходит при употреблении мужчинами банту местных сортов пива, хранящегося в негальванизированных стальных бочках, которое, в соответствии с местными обычаями, принято запивать свежей кровью животных. При гемохроматозе увеличивается концентрация железа в сыворотке крови, повышается степень насыщения трансферрина железом и увеличивается содержание ферритина в сыворотке крови. Резко возрастает содержание депонированного железа в организме. Если у здоровых людей объем депонированного железа составляет 1-3 г, то при гемохроматозе запасы депонированного железа возрастают до 20-40 г. Железо откладывается в виде водорастворимого ферритина, а также нерастворимого в воде гемосидерина. Увеличивается содержание в клетках паренхиматозных органов и кожи меланина и липофусцина. Это приводит к развитию пигментных дистрофий и повреждению клеток. Накопление в клетках железа также способствует повреждению их вследствие «окислительного» стресса:
Fe2+ +H2O2®Fe3+ + OH. + OH- Fe3+ + O2-.®Fe2+ + O2 Увеличенное образование АФК приводит к повреждению ДНК, нарушению целостности клеточных мембран и процессов пролиферации клеток, приводя вначале к обратимому повреждению клеток, а затем – к необратимому. При накоплении Fe3+ в лизосомах печени возникает дистрофия этих клеток, часть гепатоцитов погибает и замещается соединительной тканью, то есть развивается цирроз печени. Гемохроматоз часто осложняется развитием гепатоцеллюлярной карциномы из-за повреждения генетического аппарата гепатоцитов АФК. Увеличение содержания железа в клетках кожи в виде гемосидерина придает ей характерный оттенок, что послужило основанием называть гемохроматоз «бронзовой болезнью». Кроме того, гемосидерин стимулирует накопление меланина в меланоцитах. Накопление железа в поджелудочной железе сопровождается гибелью части b-клеток островков Лангерганса и уменьшением выработки инсулина. Страдает также экзокринная функция поджелудочной железы. При увеличении содержания ферритина, гемосидерина и липофусцина в сердце (особенно в кардиомиоцитах желудочков сердца) возникает фиброз и кардиомегалия; появляются признаки сердечной недостаточности. Часто у таких пациентов возникают фатальные аритмии. Избыточные количества железа откладываются также в эндокринных железах – чаще в гипофизе, надпочечниках, щитовидной железе и паращитовидных железах. Особенно часто поражается гипофиз. Характерным проявлением такого нарушения является уменьшение продукции гонадотропных гормонов, что приводит к нарушению половой функции у мужчин. При гемохроматозе также поражаются суставы. Гемосидерин накапливается в синовиальной полости сустава и хрящах, что приводит к развитию артропатий и артритов. Накопление железа в виде ферритина в макрофагах способствует ослаблению механизмов фагоцитоза. Поэтому пациенты, страдающие гемохроматозом, подвержены различным инфекционным заболеваниям, особенно вызванными Vibrio vulnificus, Listeria monocytogenes, Yersinia enterocolitica, Salmonella enteridis, Klebsiella pneumoniae, Escherichia coli и Rhizopus arrhizus. Патогенетические принципы лечения гемохроматоза основаны на ограничении потребления пищи с высоким содержанием железа и аскорбиновой кислоты, регулярных кровопусканиях, а также использовании препаратов, образующих хелатные комплексы с железом и выводящих его избыток из организма.
Методическую разработку подготовила доцент кафедры патофизиологии, к.м.н. Беляева Л.Е.
Кафедра патофизиологии ВГМУ Методическая разработка для студентов к лабораторному занятию по теме:
ПАТОФИЗИОЛОГИЯ СИСТЕМЫ КРОВИ
В12 ДЕФИЦИТНАЯ АНЕМИЯ Дефицит витамина B12 является довольно распространенным состоянием. По разным оценкам, от него страдают от 1,5% до 15% людей во всем мире.
Общие сведения о метаболизме и роли витамина В12 (цианокобаламина) в организме человека Витамин В12 поступает в организм человека с пищей, он содержится в продуктах животного происхождения – мясе, печени, почках, яичном желтке, сыре, молоке. В небольшом количестве цианокобаламин синтезируется нормальной микрофлорой кишечника, но не может в полной мере всасываться. Поэтому для обеспечения потребности организма цианокобаламин должен поступать с пищей. В пище витамин В12 связан с белком. В желудке под действием соляной кислоты и протеолитических ферментов витамин В12 высвобождается из пищи. Далее витамин В12 (внешний фактор Касла) поступает в 12-перстную кишку и соединяется с гастромукопротеином (внутренний фактор Касла), поступившим сюда из желудка. Гастромукопротеин вырабатывается париетальными клетками в фундальной части желудка. Внутренний фактор Касла защищает витамин В12 от разрушающего действия протеолитических ферментов. Далее комплекс «витамин В12 + гастромукопротеин» продвигается по тонкому кишечнику и поступает в подвздошную кишку, после чего расщепляется и через клетки слизистой оболочки всасывается в кровь, где соединяется с транспортным белком – транскобаламином. В конечном итоге витамин В12 поступает в костный мозг и печень. В костном мозге витамин В12 используется для кроветворения, в печени – депонируется и в дальнейшем поступает при необходимости в кровь. Незначительное количество витамина В12 (около 1 %) всасывается в желудке без участия внутреннего фактора. При полноценном питании суточный рацион человека содержит до 30 мкг витамина В12. Суточная потребность в витамине составляет 2-7 мкг. В организме здорового человека содержится около 2-5 мг витамина В12. Запасов витамина В12 в печени хватает на 3-5 лет после прекращения его всасывания. Свою биологическую роль витамин В12 выполняет в виде двух коферментов – метилкобаламина и 5-дезоксиаденозилкобаламина. Метилкобаламин участвует в обеспечении нормального размножения, развития и созревания клеток системы кроветворения, прежде всего эритробластического ростка и эпителия желудочно-кишечного тракта. Тетрагидрофолиевая кислота, образующаяся при участии метилкобаламина, необходима для синтеза 5,10-метилтетрагидрофолиевой кислоты (коферментная форма фолиевой кислоты), участвующей в образовании тимидинфосфата. Последний включается в ДНК эритрокариоцитов и других интенсивно делящихся клеток. Недостаток тимидинфосфата обусловливает нарушение синтеза и структуры ДНК, что приводит к нарушению синхронности между созреванием цитоплазмы и ядра. В результате в периферической крови появляются макроциты (мегалоциты), эритроциты с остатками ядерного вещества (тельца Жолли) или ядерной оболочки (кольца Кабо) и гиперсегментированные нейтрофилы. Неэффективный эритропоэз приводит к гемолизу в костном мозге и высвобождению лактатдегидрогеназы. В костном мозге разрушается до 50% макроцитов (в норме – около 20%). В связи с этим существенно снижается количество эритроцитов и в периферической крови, иногда до 0,7 – 0,8×1012 /л., а цветовой показатель увеличивается (гиперхромная анемия).
Таким образом, кофермент витамина В12 метилкобаламин необходим для нормального синтеза ДНК. Вторая метаболически активная форма витамина В12 – 5-дезоксиаденозилкобаламин регулирует синтез жирных кислот, катализируя образование янтарной кислоты из метилмалоновой. Дефицит 5-дезоксиаденозилкобаламина обусловливает нарушение образования миелина, оказывает прямое повреждающее действие на нейроны головного и спинного мозга (особенно задних и боковых его столбов), что проявляется психическими расстройствами (бред, галлюцинации), признаками фуникулярного миелоза (шаткая походка, парестезии, болевые ощущения, онемение конечностей). Таким образом, кофермент витамина В12 – 5-дезоксиаденозилкобаламин необходим для нормального метаболизма миелина нервных волокон. Кроме этих функций витамин В12 играет большую роль в обмене гомоцистеина – серосодержащей аминокислоты, которая является промежуточным продуктом обмена аминокислот метионина и цистеина. Этиология В12-дефицитной анемии Основные причины развития В12-дефицитной анемии: 1. Нарушение секреции желудком гастромукопротеина: · Атрофический аутоиммунный гастрит является наиболее частой причиной развития В12-дефицитной анемии. Такая анемия обозначается как пернициозная, или анемия Аддисона-Бирмера. Это заболевание аутоиммунной природы, сопровождается ахилией, отсутствием пепсина в желудочном соке и продукцией антител к париетальным клеткам, вырабатывающим гастромукопротеин. Часто сочетаются с пернициозной анемией и другие аутоиммунные заболевания, в особенности заболевания щитовидной железы и сахарный диабет 1 типа. · Тотальная гастрэктомия или субтотальная резекция желудка – при этом выпадает секреция гастромукопротеина. В связи с большим запасом витамина В12 в печени, В12-дефицитная анемия после гастрэктомии развивается через 4-5 лет. · Врожденное нарушение секреции гастромукопротеина. В этом случае секреция пепсина и соляной кислоты нормальная, в отличие от пернициозной анемии антитела к париетальным клеткам желудка и гастромукопротеину не выявляются. · Рак желудка, сопровождающийся атрофией слизистой оболочки желудка и прекращением продукции гастромукопротеина.
· Токсическое действие высоких доз алкоголя на слизистую желудка. 2. Нарушение всасывания витамина В12 в тонком кишечнике: · Резекция участка подвздошной кишки (более 60 см); · Синдром мальабсорбции различного генеза (ферментные энтеропатии, целиакия, тропическое спру, болезнь Крона, энтериты); · Нарушение всасывания витамина В12, вызванное приемом лекарственных средств (колхицин, неомицин, бигуаниды, ПАСК и др.). 3. Конкурентное расходование витамина В12 наблюдается при инвазии широким лентецом, который, обитая в тонком кишечнике, поглощает поступающий сюда витамина В12. 4. Повышенный расход витамина В12, что может наблюдаться при многоплодной беременности. У ребенка, рожденного матерью с дефицитом В12 и находившегося исключительно на грудном вскармливании, может также наблюдаться дефицит витамина В12. 5. Нарушение поступления витамина В12 с пищей наблюдается редко у лиц, которые являются строгими вегетарианцами и не употребляют в пищу продукты животного происхождения. 6. Снижение запасов витамина В12 может наблюдаться при циррозе печени. 7. Нарушение транспорта витамина В12, связанное с количественным или функциональным дефицитом транскобаламина. Патогенез В12-дефицитной анемии Таблица 1 Патогенез важнейших системных проявлений при дефиците витамина В12
Диагностика и выявление причины дефицита витамина В12 Определение витамина В12 в сыворотке крови - самый обычный метод диагностики его дефицита, однако имеющиеся методы недостаточно специфичны. Как правило, низкие показатели (менее 150 пг/мл) убедительно свидетельствуют о дефиците В12. При пограничных результатах (от 150 до 250 пг/мл) эти определения должны быть дополнены клиническими данными и другими тестами. Дефицит витамина В12 в тканях приводит к повышению уровня ММК (метилмалоновой кислоты) и гомоцистеина в сыворотке крови, поэтому измерение их уровня можно использовать при диагностике дефицита В12. Уровень ММК и гомоцистеина заметно повышается у подавляющего большинства (более 89%) пациентов с дефицитом витамина В12, включая тех, у которых имеется только неврологическая симптоматика (т.е. нет признаков анемии). После начала лечения уровень ММК и гомоцистеина снижается, что может быть использовано для отражения адекватности заместительной терапии. Повышение уровня ММК – достаточно специфический показатель дефицита витамина В12. Умеренное повышение уровня ММК (от 300 до 700 нмоль/л) может отмечаться и при почечной недостаточности. Однако при дефиците витамина В12 уровень ММК достигает 500 – 1000 нмоль/л. Уровень сывороточного гомоцистеина является менее специфичным показателем, т.к. он повышается не только при дефиците витамина В12, но и при дефиците фолиевой кислоты. После установления наличия дефицита витамина В12, необходимо выяснить его причину. У большинства больных причиной является нарушение синтеза гастромукопротеина вследствие развития атрофического гастрита. Диагноз пернициозной анемии устанавливается при обнаружении в крови антител к париетальным клеткам желудка или гастромукопротеину. Другие причины дефицита витамина В12 устанавливаются путем тщательного анализа клинической картины заболевания. Практический врач всегда должен помнить о том, что мегалобластная анемия может сочетаться с раком желудка. Поэтому всем больным с мегалобластной анемией необходимо обязательно проводить фиброгастроскопию и биопсию слизистой оболочки желудка для исключения рака желудка. У всех больных с В12-дефицитной анемией необходимо произвести исследование кала для исключения инвазии широким лентецом. Следует выяснить также, производилась ли больному операция на желудке. В12-дефицитная анемия развивается через 3 – 5 лет после гастрэктомии в связи с отсутствием секреции гастромукопротеина. Принципы лечения дефицита витамина В12 Коррекция дефицита витамина В12 осуществляется с учетом этиологии, выраженности анемии, наличия неврологических нарушений посредством назначения пероральных или внутримышечных его препаратов (в случае невозможности его всасывания в ЖКТ).
Методическая разработка составлена ассистентом кафедры И.В. Лигецкой СИНДРОМ ДИССЕМИНИРОВАННОГО ВНУТРИСОСУДИСТОГО СВЕРТЫВАНИЯ (ДВС-синдром)
Определение. Синдром диссеминированного внутрисосудистого свертывания (ДВС-синдром) - приобретенный синдром, характеризующийся внутрисосудистой активацией свертывания крови без определенной локализации под влиянием различных факторов. Повреждение сосудов микроциркуляторного русла может быть как причиной, так и следствием этого синдрома, и в тяжелых случаях способно привести к полиорганной недостаточности. (Комиссия Комитета по науке и стандартизации Международного Общества Гемостаза и Тромбоза, 2001 г.)
Эпидемиология ДВС-синдрома. Частота встречаемости среди пациентов, находящихся в стационаре – 1:1000. Смертность – 30-80%.
Этиология ДВС-синдрома: ü инфекции, сепсис; ü акушерские осложнения; ü шок любой этиологии; ü злокачественные новообразования; ü травмы, ожоги, отравления, укусы змей; ü острый панкреатит, почечная недостаточность.
Виды ДВС-синдрома в зависимости от характера течения: · острый · подострый · хронический
Фазы развития ДВС-синдрома: 1. активация системы гемостаза 2. фаза гиперкоагуляции 3. переходная фаза 4. коагулопатия потребления 5. фаза исходов
ГЕМОДИНАМИЧЕСКИЕ ИЗМЕНЕНИЯ ПРИ ПРИОБРЕТЁННЫХ ПОРОКАХ СЕРДЦА
Виды приобретённых пороков сердца: 1. Недостаточность митрального клапана (НМК). 2. Стеноз левого атриовентрикулярного отверстия (митральный стеноз) (МС). 3. Недостаточность аортального клапана (аортальная недостаточность) (АН). 4. Стеноз устья аорты (аортальный стеноз) (АС). 5. Недостаточность трикуспидального клапана (трикуспидальная недостаточность) (ТН). 6. Стеноз правого атриовентрикулярного отверстия (трикуспидальный стеноз) (ТС). 7. Недостаточность клапана легочного ствола (НКЛС), 8. Стеноз устья легочного ствола (СУЛС). При НМК имеет место систолическая регургитация крови (ретроградный ток крови) из левого желудочка (ЛЖ) в левое предсердие (ЛП) и последующий увеличенный выброс крови из ЛП в ЛЖ. Это приводит к увеличению нагрузки объёмом ЛП и ЛЖ и развитию изотонической гиперфункции и эксцентрической гипертрофии этих отделов сердца. При МС увеличивается сопротивление тока крови из ЛП в ЛЖ, что приводит к развитию изометрической гиперфункции и концентрической гипертрофии ЛП. При АН имеет место диастолическая регургитация крови из аорты в ЛЖ. Это приводит к перегрузке ЛЖ объёмом крови, развитию изотонической гиперфункции и эксцентрической гипертрофии ЛЖ. При данном пороке сердца увеличивается диастолическое наполнение ЛЖ, возрастает систолический выброс (СВ) и минутный объём крови (МОК), увеличивается систолическое (САД) и пульсовое (ПАД) артериальное давление, снижается диастолическое (ДАД) артериальное давление. Пульс при этом пороке высокий (altus), быстрый (celer), частый (freguens). Увеличение САД – следствие возрастания СВ. Регургитация крови приводит к снижению ДАД. При АС возрастает сопротивление кровотоку в суженном устье аорты. ЛЖ при этом подвергается перегрузке сопротивлением (давлением), что приводит к развитию изометрической гиперфункции и концентрической гипертрофии ЛЖ. Пульс при этом пороке малый (parvus), медленный (tardus), рудкий (rarus). САД несколько снижается, ДАД увеличивается, ПАД уменьшается. При всех указанных пороках может развиться недостаточность левого сердца с характерными изменениями. При ТН имеет место регургитация крови из правого желудочка (ПЖ) в правое предсердие (ПП), что приводит к перегрузке ПЖ и ПП объёмом и развитию изотонической гиперфункции и эксцентрической гипертрофии ПП и ПЖ. При ТС увеличивается сопротивление току крови из ПП и ПЖ. Вследствие перегрузки сопротивлением ПП развивается изометрическая гиперфункция и концентрация гипертрофии ПП. При НКЛС имеет место регургитация крови из легочного ствола в ПЖ. ПЖ подвергается перегрузке объёмом, развивается изотоническая гиперфункция и эксцентрическая гипертрофия этого отдела сердца. При СУЛС возрастает сопротивление току крови из ПЖ в легочной ствол, это приводит к развитию изометрической гиперфункции и концентрической гипертрофии ПЖ.
ИЗМЕНЕНИЯ ДЕЯТЕЛЬНОСТИ СЕРДЕЧНО-СЛСУДИСТОЙ СИСТЕМЫ (ССС) ПРИ ПОВРЕЖДЕНИИ СИСТЕМЫ ВНЕШНЕГО ДЫХАНИЯ (СВД) Существенные изменения деятельности ССС имеют место при обструктивных и рестриктивных поражениях СВД. Обструктивный тип характеризуется сужением бронхов, рестриктивный тип – уменьшением растяжимости легочной ткани. При обоих формах поражения СВД имеет место повышение сосудистого сопротивления (СС) в лёгких. Увеличение СС в лёгких может быть следствием функциональных и структурных изменений. Функциональные изменения проявляются повышением тонуса резистивных сосудов лёгких. Одним из патогенетических факторов повышения тонуса сосудов является рефлекс Эйлера Лильестранда. Рефлекс возникает в ответ на уменьшение парциального давления кислорода в альвеолах. Указанная реакция может быть локальной и генерализованной. Локальная реакция, характеризующаяся уменьшением притока крови к участкам лёгких с низким парциальным давлением кислорода в альвеолах, имеет адаптивное значение. Она способствует лучшему газообмену между альвеолярным воздухом и протекающей через сосуды лёгких кровью. Благодаря указанной реакции кровь в основном направляется в те отделы лёгких, где имеет место большое содержание кислорода. Генерализованная реакция Эйлера Лильестранда носит патологический характер, приводит к существенному увеличению СС в лёгких. Структурные изменения проявляются увеличением числа функционирующих сосудов. Увеличение СС в лёгких приводит к увеличению нагрузки сопротивлением на ПЖ. Вследствие этого развивается изометрическая гиперфункция и концентрическая гипертрофия ПЖ, которая может привести к недостаточности правого сердца. Указанные изменения получили название «Легочное сердце». ПАТОГЕНЕЗ ОСТРОГО ИШЕМИЧЕСКОГО И РЕПЕРФУЗИОННОГО ПОВРЕЖДЕНИЯ МИОКАРДА
Активное клиническое применение тромболитических препаратов и методов эндоваскулярных вмешательств при остром коронарном синдроме позволило снизить смертность от инфаркта миокарда, но привело к осознанию того, что восстановление кровотока в ишемизированном миокарде вызывает дополнительное повреждение сердечной мышцы. Реперфузионное повреждение миокарда требует понимания его механизмов и внедрения в клиническую практику новых способов защиты.
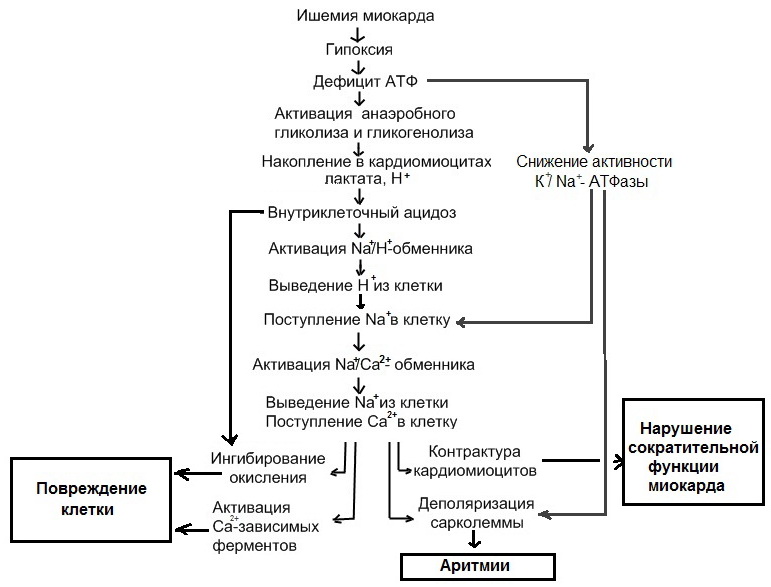
Патогенез ишемического повреждения миокарда Чаще всего в основе развития ишемического повреждения миокарда лежит нарушение притока артериальной крови по одной коронарных артерий, возникающее чаще всего в результате тромбоза артерии в месте разрыва атеросклеротической бляшки.
При ишемии наиболее ранние изменения возникают в системе энергетического метаболизма кардиомиоцитов. Так, транспорт электронов по дыхательной цепи митохондрий и образование АТФ путем окислительного фосфорилирования прекращаются в течение нескольких секунд после наступления полной ишемии. Практически одновременно происходит активация ключевых ферментов гликогенолиза (фосфорилазы) и гликолиза (фосфофруктокиназы). Дополнительная активация фосфорилазы осуществляется за счет высвобождения катехоламинов из ишемизированных нервных окончаний и стимуляции β-адренорецепторов кардиомиоцитов с последующим возрастанием внутриклеточной концентрации циклического аденозинмонофосфата. В итоге гликоген включается в гликолитический путь. Запасы высокоэнергетических фосфатов, среди которых важнейшую роль играет АТФ, в миокарде невелики. При острой ишемии потребность в АТФ многократно превышает скорость ее образования, и единственным ее источником при этом является гликолиз. Гликолитическая продукция АТФ также быстро снижается вследствие накопления в миокарде конечных продуктов гликолиза - протонов, лактата и НАДН. Отсутствие кислорода и торможение гликолиза являются не единственными причинами снижения уровня АТФ в ишемизированном миокарде. Снижению АТФ при ишемии также способствует нарушение ее транспорта из митохондрий и активация некоторых циклических метаболических процессов, в ходе которых происходит усиленная утилизация АТФ. К таким процессам относится избыточный захват кальция (Са2+) в саркоплазматический ретикулум (СР) и его высвобождение из СР, а также распад триглицеридов до свободных жирных кислот с последующим их ресинтезом. Эти процессы усугубляют и без того выраженный энергетический дефицит клеток миокарда. Метаболические нарушения в ишемизированном миокарде затрагивают также обмен липидов и белков. При ишемии средней тяжести, в частности, при наличии коллатерального кровотока, происходит усиление захвата жирных кислот (ЖК) в зоне ишемии. Скорость захвата ЖК при этом превышает скорость их утилизации, что приводит к накоплению избытка свободных ЖК, ацил-КоА и ацилкарнитина, которые в высоких концентрациях обладают выраженным повреждающим действием в отношении функций сарколеммы и митохондрий. При тяжелой ишемии происходит полное угнетение митохондриального окисления ЖК и единственным источником энергии служит анаэробный метаболизм глюкозы. Наступление лактат-ацидоза и гибель клетки при этом происходят значительно быстрее. Важнейшим следствием нарушений энергетического метаболизма миокарда в ранней стадии ишемии является его сократительная дисфункция, возникающая в результате:
В раннем периоде ишемии происходит возрастание внутриклеточной концентрации Са2+, вследствие его поступления по потенциал-зависимым Са2+ каналам, выхода его из СР, а также возрастания его притока в результате активации «обратного» варианта работы Na+/Ca2+обменника (выведение Na+ из клетки в обмен на поступающий внутрь нее Са2+). Данный вариант работы Na+/Ca2+обменника активируется в ходе ишемии вследствие повышения внутриклеточной концентрации Na+, поступающего в клетку в обмен на протоны, выводимые Na+/H+ насосом для компенсации внутриклеточного ацидоза. Повышение внутриклеточной концентрации Са2+ является ключевым звеном патогенетической цепи, ведущей к необратимому повреждению кардиомиоцитов, т.к. приводит активации Са2+-зависимых протеаз, липаз и фосфолипаз, ингибированию биологического окисления в митохондриях и их повреждению, деполяризации сарколеммы и ишемической контрактуре кардиомиоцитов.
Критическим событием в патогенезе ишемического повреждения миокарда является переход обратимой стадии повреждения в необратимую. Последняя характеризуется гибелью кардиомиоцитов в результате некроза и апоптоза. Важнейшим механизмом необратимого повреждения кардиомиоцитов является нарушение целостности их мембраны посредством следующих механизмов:
Распространение зоны некроза в ишемизированном миокарде происходит в виде «волны клеточной гибели», зарождающейся в субэндокарде и постепенно, с течением времени, распространяющейся по направлению к эпикардиалъной поверхности. Это объясняется тем, что внутренние слои миокарда наиболее удалены от проходящих субэпикардиально коронарных артерий и, кроме того, подвергаются наибольшему механическому сдавлению в систолу.
МЕХАНИЗМЫ РЕПЕРФУЗИОННОГО ПОВРЕЖДЕНИЯ МИОКАРДА
Раннее восстановление кровотока в ишемизированном миокарде (реперфузия) является оптимальным методом борьбы с ишемическим повреждением миокарда. Реперфузия миокарда после кратков
|
|||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2021-07-18; просмотров: 66; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.188.29.73 (0.091 с.) |