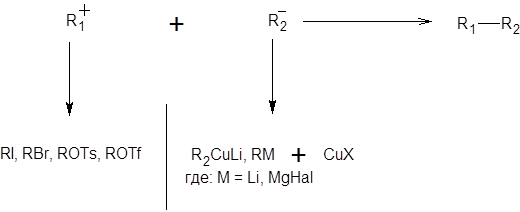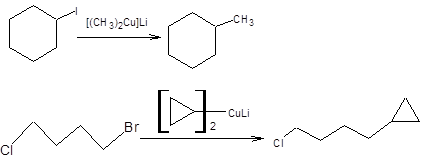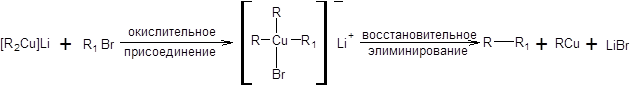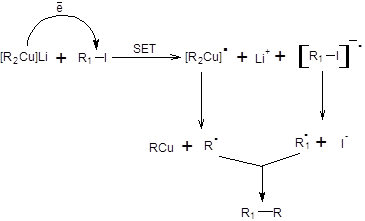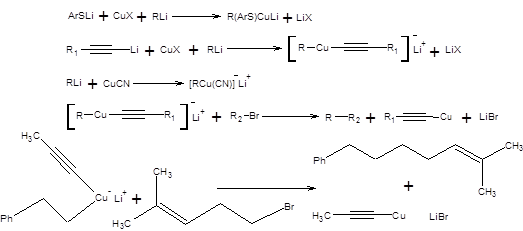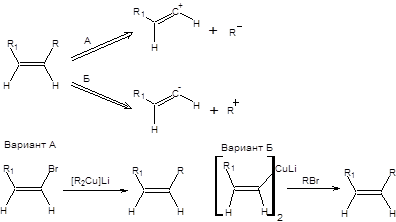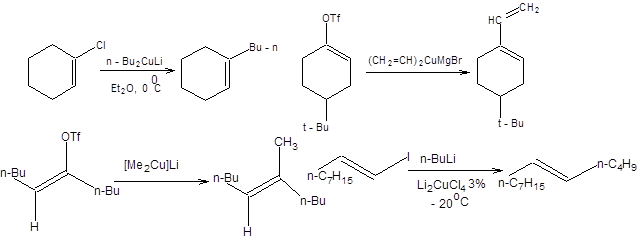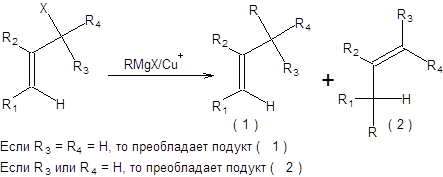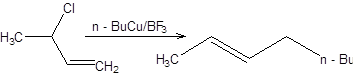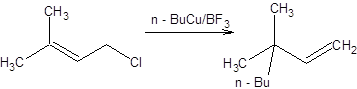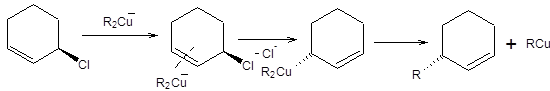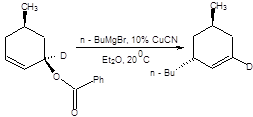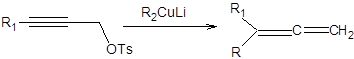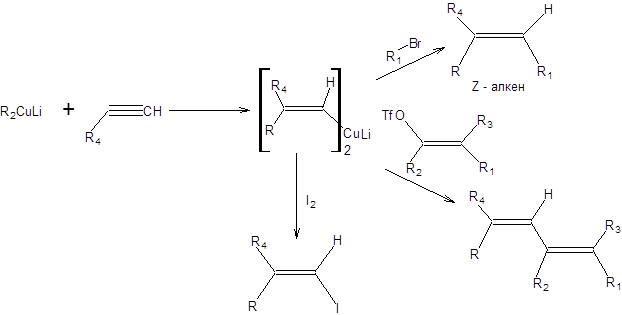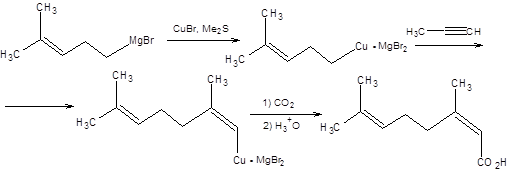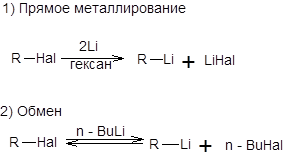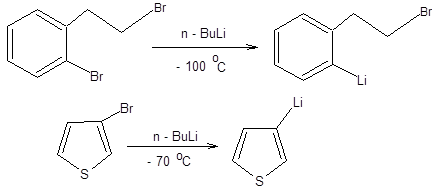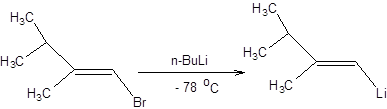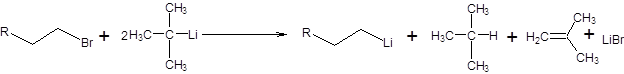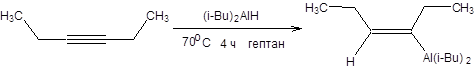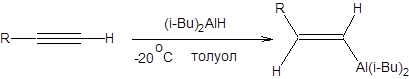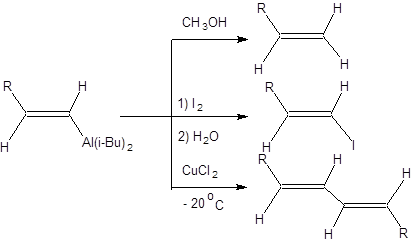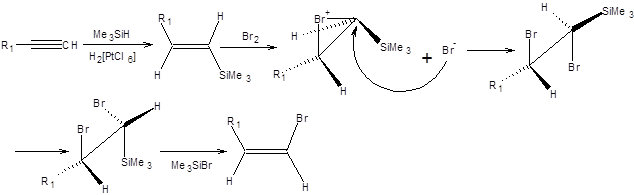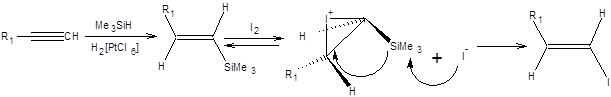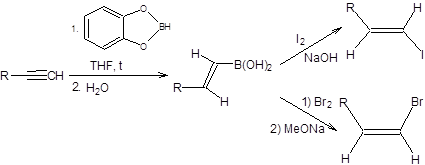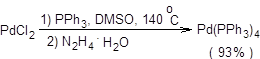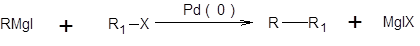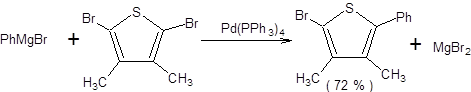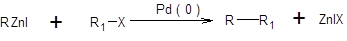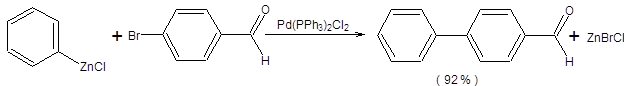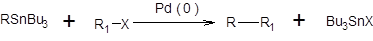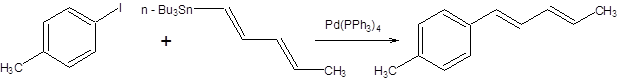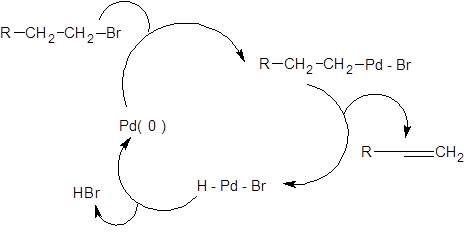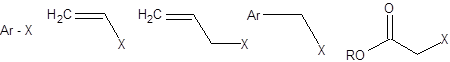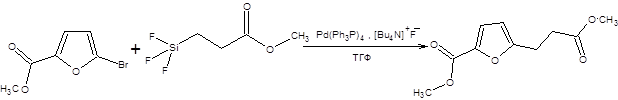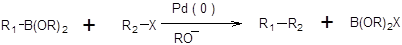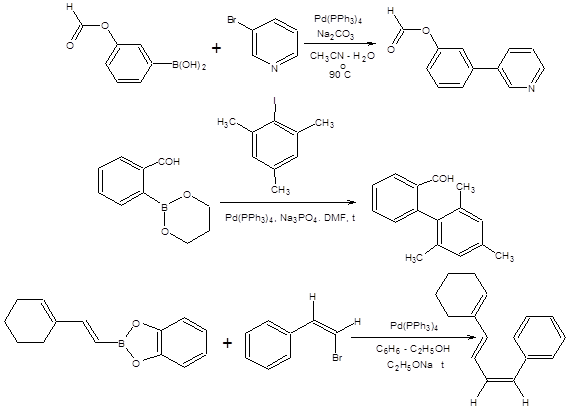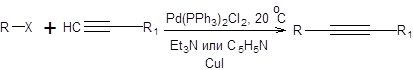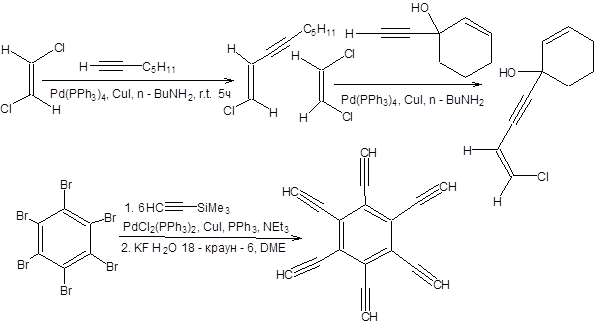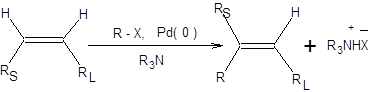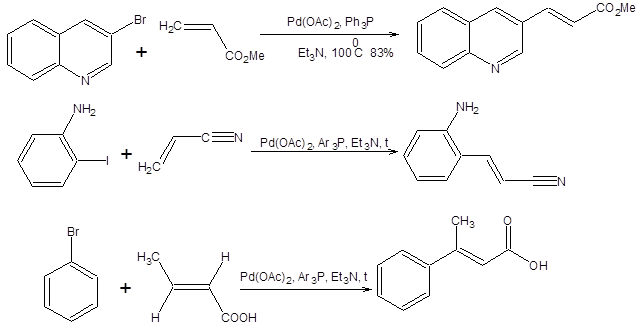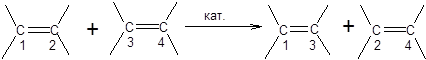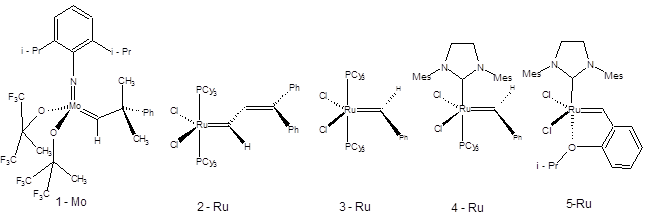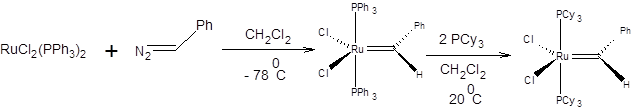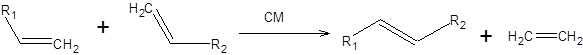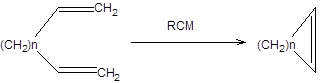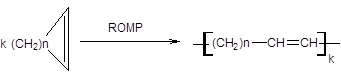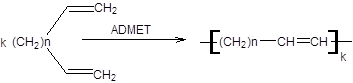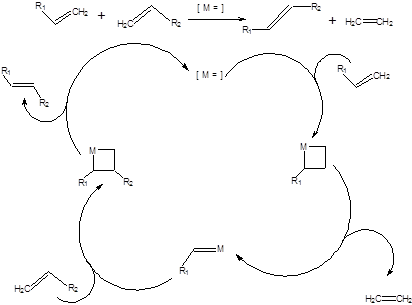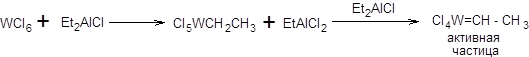Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Применение купратов в синтезе алкенов
Использование купратов в органическом синтезе открыло принципиально новые возможности для эффективного построения С–С-связи, благодаря их относительно низкой основности, высокой нуклеофильности, а также широкому разнообразию комплексов меди, отличающихся числом и природой лигандов. Широкий ассортимент используемых купратов позволяет осуществлять сборку С–С-связей самых различных типов, а также подбирать оптимальные условия для реализации необходимой синтетической трансформации [1]. Список широко употребляемых в синтетической практике органических купратов приведен в таблице.
Таблица
Используя перечисленные реагенты, возможно осуществить сборку углерод–углеродных связей между sp3 – sp3 и sp3 – sp2 - гибридными атомами углерода. 1. Построение С – С-связи между sp 3 - sp 3 - гибридными атомами углерода Для построения С–С-связи между sp3 – sp3 - гибридными атомами углерода с успехом применяются гомокупраты, смешенные купраты и гетерокупраты. Гомокупраты получают обработкой одного эквивалента галогенида меди двумя эквивалентами литийорганического соединения, как показано на схеме 3.1.
Схема 3.1
Гомокупраты взаимодействуют с бромпроизводными, йодпроизводными, трифлатами и тозилатами с образованием новой углерод-углеродной связи с высоким выходом по эквиваленту карбкатиона (схема 3.2) [1, 2].
Схема 3.2
Механизм взаимодействия купратов с галогенопроизводными сложен и до конца не установлен, кроме того он может изменяться в зависимости от природы формального нуклеофуга. Известно, что взаимодействие бромпроизводных и тозилатов с купратами происходит с обращением конфигурации электрофильного центра, тогда как реакции с участием йодалканов приводят к рацемизации, что может свидетельствовать в пользу радикального механизма в последнем случае. Для бромпроизводных и тозилатов в качестве рабочей гипотезы можно допустить механизм, включающий окислительное присоединение галогеналкана и последующее восстановительное элиминирование продукта реакции, как показано на схеме 3.3.
Схема 3.3
В случае участия йодпроизводных вероятно возможен ион-радикальный механизм, объясняющий рацемизацию хирального электрофильного углеродного центра (схема 3.4).
Схема 3.4
Применение гомокупратов в синтезе эффективно для переноса малых фрагментов, соответствующих легко доступным предшественникам, так как образующаяся наряду с целевым продуктом алкилмедь не вступает в реакцию и быстро разлагается. Поэтому выход реакции по нуклеофильной компоненте не может превышать 50% в случае использования гомокупратов. Вместе с тем часто необходимо использование малодоступных эквивалентов нуклеофильных синтонов в синтезе, в этом случае использование гомокупратов очевидно не целесообразно. Для устранения указанного затруднения эффективно использование смешанных купратов, содержащих вспомогательные группы, не участвующие в реакции, а также гетерокупратов. В качестве вспомогательных фрагментов эффективны ацетиленидные группы смешанных купратов, а также сульфидный фрагмент гетерокупратов. Примеры синтеза смешанных купратов и гетерокупратов, а также некоторые реакции с их участием представлены на схеме 3.5.
Схема 3.5
2. Построение С–С-связи между sp 3 – sp 2 -гибридными атомами углерода В терминах ретросинтетического анализа расчленение С–С-связи между sp3 – sp2 гибридными атомами углерода можно реализовать двумя различными способами (схема 3.6).
Схема 3.6
Способ А) (схема 3.6) предполагает использование винильного фрагмента в качестве эквивалента электрофильного синтона. Способ Б) (схема 3.6) предполагает перенос винильного фрагмента в роли нуклеофильного синтона. Оба подхода в равной степени эффективны для построения С – С-связи между sp3 – sp2-гибридными атомами углерода и используются в современном органическом синтезе. Некоторые типичные примеры применения купратов в создании С – С-связи между sp3 – sp2 -гибридными атомами углерода приведены на схеме 3.7. [1, 3, 4].
Схема 3.7
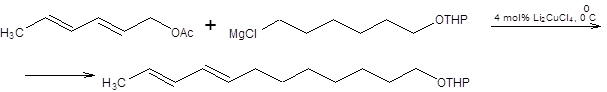
3. Синтез алкенов построением С–С-связи между sp 3 – sp 3 -гибридными атомами углерода с участием аллильных систем Использование аллилгалогенидов и сложных эфиров аллиловых спиртов в реакциях с алкилкупратами открывает перспективный путь к синтезу алкенов, причем в зависимости от типа используемого аллильного субстрата и природы купрата возможна реализация одного из двух альтернативных направлений замещения [5]. Первое направление алкилирования можно рассматривать как формальное SN2 замещение, второе направление можно рассматривать как SN2’замещение, сопровождающееся перемещением двойной связи с одновременным выбросом нуклеофуга. Сходство рассматриваемых процессов с классическими реакциями нуклеофильного замещения ограничено лишь формальным результатом – продуктами реакции, тогда как механизм реализации замещения принципиально различен. Общая схема взаимодействия купратов с аллильными электрофилами приведена ниже:
Схема 3.8
Реакция обыкновенно проводится смешением аллильных электрофилов с литийорганическими или магнийорганическими соединениями в присутствии солей меди (I). В качестве эквивалентов Сu+ используются галогениды меди в эквимоляном соотношение или даже в меньшем количестве, а также Li2CuCl4 – эффективный в каталитических количествах. Направление замещения критическим образом зависит от структуры аллильного электрофила, а также природы противоиона купрата. Если в качестве субстратов используются первичные аллильные электрофилы, а литийорганические или магнийорганические соединения в качестве нуклеофилов в присутствии каталитических количеств перхлората лития, то основным направлением реакции будет формальное SN2 замещение, как показано на схеме 3.9.
Схема 3.9. В случае использования вторичных и третичных аллильных электрофилов преобладают продукты аллильной перегруппировки. Полного преобладания продуктов аллильной перегруппировки даже в случае первичных аллильных электрофильных субстратов можно достичь, применяя систему RCu . BF3. Система RCu . BF3 образуется при смешении соответствующего алкиллития, йодида меди и эфирата трехфтористого бора в эквимолярном соотношении (схема 3.10). RLi + CuI + BF3.Et2O = RCu .BF3 + LiI + Et2O
Схема 3.10
Высокой эффективностью обладают реагенты [R2Cu]ZnCl, которые обеспечивают высокую селективность образования продукта аллильной перегруппировки, а также являются диастереоселективными реагентами, причем вводимая алкильная группа, всегда занимает анти-положение по отношению к нуклеофугу. Механизм данной реакции, объясняющий ее диастереоселективность, предполагает анти-координацию купратного реагента по двойной связи аллильного реагента с последующим элиминированием нуклеофуга с синхронным перемещением двойной связи. На завершающей стадии происходит восстановительное элиминирование с образованием продукта реакции и выделением алкилмеди (схема 3.11).
Схема 3.11
При использовании пропаргиловых электрофилов в реакциях с купратами всегда образуются продукты алленового типа, соответствующие формальному SN2’замещению, как показано на схеме 3.12.
Схема 3.12
Реакция карбокуприрования Литийорганические соединения и реактивы Гриньяра являются сильными основаниями и образуют соли при взаимодействии с терминальными алкинами (схема 3.13).
Схема 3. 13
Органические купраты почти также нуклеофильны, как реактивы Гриньяра, но существенно менее основны. Ацетилены в присутствие органокупратов не ведут себя как протонные кислоты, а вступают в реакцию формального нуклеофильного присоединения (реакцию карбокуприрования). Результатом реакции карбокуприрования является удлинение углеродной цепи на два атома углерода с образованием эквивалента карбаниона, который может взаимодействовать с широким кругом электрофилов, как показано на схеме 3.14.
Схема 3.14
Реакция карбокуприрования стереоселективна. Присоединение органокупратов протекает как син-присоединение по тройной связи ацетиленов, что открывает путь к синтезу алкенов и диенов заданной конфигурации. Если в реакцию вступают терминальные алкины, отличные от ацетилена, то новая алкильная группа вступает к уже имеющейся в субстрате группе, что отвечает образованию наиболее устойчивого карбаниона. Реакция карбокуприрования позволяет осуществлять стереоконтроль при синтезе тризамещенных алкенов. На схеме 3.15 представлен синтез (Z)-гераниевой кислоты как иллюстрация применения реакции карбокуприрования в синтезе природных соединений, содержащих тризамещенную двойную связь заданной конфигурации.
Схема 3.15 Реакции кросс-сочетания Синтез алкенов и диенов заданной конфигурации может быть эффективно осуществлен посредством взаимодействия винилгалогенидов и металлорганических соединений самого различного строения, в присутствии каталитических количеств комплексов палладия. Особое значение приобрели реакции с участием органических соединений кремния, бора, олова, цинка, магния и алюминия. Органические производные перечисленных элементов получают из соответствующих литийорганических соединений обычными способами, например, взаимодействием с галогенидами этих элементов. Литийорганические соединения получают исходя из галогенопроизводных прямым литированием или обменом с использованием широко доступного битиллития, как показано на схеме 3.16.
Схема 3.16
В случае синтеза литийорганических соединений методом обмена необходимо учитывать обратимость этого процесса, причем равновесие смещено в сторону литийорганического соединения, соответствующего более устойчивому карбаниону. Обмен галогена на литий происходит эффективно для бром- и йодпроизводных, тогда как хлорпроизводные малоактивны. Так как стабильность карбанионов увеличивается при переходе от sp3- к sp- гибридизованным углеродным центрам, то следует ожидать почти полного смещения равновесия реакции обмена в сторону образования винильных и арильных литийорагических соединений при взаимодействии винилгалогенидов и арилгалогенидов с коммерчески доступным н-бутиллитием (схема 3.17).
Схема 3.17
Для успешного проведения обмена галогена с участием первичных галогенопроизводных удобным реагентом является, трет-бутиллитий, который в целях доведения реакции до конца используют в количестве двух эквивалентов в расчете на один эквивалент галогенопроизводного (схема 3.18).
Схема 3.18 Магний- и цинкорганические соединения часто получают прямым синтезом из соответствующих металлов и галогенопроизводных. Цинкорганические соединения также получают обработкой литийорганических и магнийорганических соединений галогендами цинка (схема 3.19).
Схема 3.20 Алюминийорганические и кремнийорганические соединения также получают взаимодействием алюминия и кремния с галогенопроизводными. Эти реакции проходят в жестких условиях с образованием сложной смеси продуктов, что сводит значимость данных реакций в тонком органическом синтезе к минимуму, несмотря на их важное место в промышленном органическом синтезе. В связи с этими затруднениями большое значение приобрели реакции гидросилилирования и гидроалюминирования. Данные реакции протекают региоселективно и стереоселективно против правила Марковникова как син-присоединение (схема 3.21).
Схема 3.21
Гидроалюминирование алкинов позволяет проводить их стереоселективное восстановление протодеметаллированием получаемых алюмийниорганических соединений, получать (E)-винилгалогениды обработкой продуктов присоединения галогенами и синтезировать симметричные (Е, E)-диены окислением как показано на схеме 3.22.
Схема 3.22 Винилгалогениды заданного строения и конфигурации получают из терминальных алкинов их гидросилилированием с образованием (Е)-винилсиланов с последующей обработкой бромом или йодом. При обработке бромом наблюдается его присоединение по двойной связи (Е)-винилсилана, с последующим элиминированием бромсилана, что проводит к образованию (Z)-изомерного винилбромида (схема 3.23).
Схема 3.23
Обработка (Е)-винилсиланов йодом приводит к образованию (Е)-винилгалогенидов, как показано на схеме 3.24.
Схема 3.24
Столь существенная разница в стереохимическом результате состоит в том, что йод присоединяется обратимо, тогда как бром необратимо. Выигрышной альтернативой гидросилилированию алкинов служит гидроборирование, протекающее с большим выходом продуктов, в более мягких условиях. Один из лучших реагентов для гидроборирования алкинов –пирокатехолборан, присоединяющийся по тройной связи с высокой региоселективностью против правила Марковникова, при повышенной температуре. Гидролиз продуктов присоединения пирокатехолборана к алкинам приводит к образованию винилбороновых кислот, занимающих чрезвычайно важное место в современном органическом синтезе, как стандартные реагенты для проведения реакции Сузуки, а также для синтеза винилгалогенидов заданной кофигурации. Синтез винилбороновых кислот из терминальных ацетиленов и их превращение в винилгалогенды заданной конфигурации приведены на схеме 3.25 [6].
Схема 3.25 Процессы с участием винилгалогенидов и арилгалогенидов, а также металлорганических соединений, приводящие к образованию новой С–С-связи в условиях металлокомплексного катализа, известны как реакции кросс-сочетания. Если в качестве металлорганической компоненты используются органические соединения кремния, бора, олова, магния и цинка, то для протекания таких реакций требуется катализатор. В качестве катализатора обычно используются комплексы никеля или палладия. Наибольшую популярность для проведения реакций кросс-сочетания получил катализатор тетракис(трифенилфосфин)палладий – (Ph3P)4Pd, который получают последовательной обработкой хлорида палладия (II) трифенилфосфином и гидразингидратом (схема 3.26).
Схема 3.26
В качестве катализаторов могут быть использованы любые комплексы Pd(0), а также соли Pd(II), которые в условиях проведения реакции восстанавливаются до каталитически активных комплексов Pd(0). В зависимости от типа используемого элементоорганического соединения различают следующие реакции кросс-сочетания: реакция Кумада:
реакция Негиши:
реакция Стилле:
Реакции Негиши, Стилле и Кумада отличаются лишь природой металлорганической компоненты, тогда как их механизм аналогичен и включает последовательность стадий окислительного присоединения, переметаллирования и восстановительного элиминирования [7–11]. Наиболее медленной стадией обычно является переметаллирование. Каталитический цикл, соответствующий реакциям Негиши, Стилле и Кумада, представлен в общем виде на схеме 3.27.
где М – (MgX, ZnX, SnR3), L - лиганд. Схема 3.27 Использование реакций кросс-сочетания в синтезе накладывает ограничения на строение формальной электрофильной компоненты. В качестве формальных электрофилов в реакциях кросс-сочетания нашли применение бромпроизводные, йодпроизводные и трифлаты. Хлорпроизводные реагируют с существенно меньшей скоростью, а в некоторых случаях не реагируют вовсе, фторпроизводные в реакции кросс-сочетания не вступают. Ограничения накладываются и на строение радикала электрофильной компоненты. Использование насыщенных алкилгалогенидов приводит к неудовлетворительным результатам вследствие высокой скорости b-элиминирования, что в итоге приводит к образованию алкена, а не желаемого продукта кросс-сочетания. Механизм b-элиминирования из насыщенных галогенопроизводных предстален на схеме 3.28.
Схема 3.28
Поэтому в качестве электрофильной компоненты реакций кросс-сочетания используются арильные, аллильные и винильные производные (схема 3.29) не содержащие атомов водорода при sр3-гибридном β-углеродном атоме.
Схема 3.30
Наиболее значимой в современном органическом синтезе из трех рассмотренных реакций кросс-сочетания является реакция Стилле, так как ее проведение сопряжено с использованием инертных по отношению к большинству функциональных групп оловоорганических соединений, что делает ее почти универсальным методом создания С–С-связи, причем эффективность протекания реакции Стилле практически не зависит от природы и числа функциональных групп в субстрате. В отличие от оловоорганических соединений, органические производные цинка и особенно магния проявляют высокую реакционную способность к ряду функциональных групп, что обозначило меньшую синтетическую значимость реакций Кумада и Негиши в органическом синтезе. Оловоорганические соединения удобны в обращении ввиду их устойчивости по отношению к влаге и воздуху. Все указанные преимущества реакции Стилле обозначили ее широкое применение в органическом синтезе, которое можно проиллюстрировать некоторыми примерами, представленными на схеме 3.31.
Схема 3.31
Оловоорганические соединения доступны из литийорганических и магнийорганических соединений путем их обработки галогенидами олова (схема 3.32).
Схема 3.32
Главным недостатком реакции Стилле является необходимость использования высокотоксичных оловоорганических соединений, что подчас заставляет обращаться к альтернативным методам проведения кросс-сочетания. Несколько отличаются от рассмотренных реакций кросс-сочетаний, реакции Хияма и Сузуки. Реакция Хияма протекает с участием трифторсилильных производных в присутствии активирующих агентов, таких как фториды или гидроксиды (схема 3.33). Роль фторидов и гидроксидов состоит в их координации по трифторсилильному фрагменту, что, в конечном счете, приводит к облегчению переметаллирования [12].
Схема 3.33
В реакции Сузуки в качестве формальной нуклеофильной компоненты используются бороновые кислоты и их производные. Реакция Сузуки отличается наличием стадии активации, которая осуществляется при участии алкоголят-иона или другого основания, облегчающего переметаллирование (схема 3.34).
Схема 3.34
Каталитический цикл реакции Сузуки включает стадии окислительного присоединения, переметаллирования, промотируемого основаниями, и восстановительного элиминирования (схема 3.35) [13,14].
Схема 3.35 Реакция Сузуки протекает медленнее, чем реакция Стилле, однако меньшая токсичность бороновых кислот и их эфиров, а также наличие значительного числа методов их стереоконтролируемого синтеза определили значимость реакции Сузуки в первую очередь как метода стереорегулируемого синтеза диенов и полиеновых структур. Следует подчеркнуть, что реакция Сузуки протекает с сохранением конфигурации для винильных и циклопропильных производных (схема 3.36).
Схема 3.36 Некоторые примеры использования реакции Сузуки приведены на схеме 3.37:
Схема 3.37 Все рассмотренные выше реакции кросс-сочетания были направлены на построение С–С-связи между двумя sр2-гибридными атомами углерода. Важную задачу в синтетической органической химии составляет построение сопряженных ениновых систем, причем их получение классическими методами составляет весьма сложную задачу, решаемую лишь разработкой многостадийных синтетических схем. В этом отношении представляет интерес метод, позволяющий решить эту задачу в одну стадию, сводя нетривиальную проблему классического органического синтеза к применению одной весьма универсальной реакции кросс-сочетания. Поэтому, особое место среди реакций кросс-сочетания занимает реакция Соногаширы, позволяющая проводить построение сопряженной ениновой системы в одну стадию в мягких условиях (схема 3.38) [15,16].
Схема 3.38
Механизм реакции Соногаширы включает два сопряженных каталитических цикла (схема 3.38). Цикл (А) включает последовательные стадии окислительного присоединения, переметаллирования и восстановительного элиминирования, тогда как цикл (Б) ответственен за образование ацетиленидов меди, которые впоследствии участвуют в реакции переметаллирования в цикле (А). В качестве акцептора выделяющегося галогеноводорода в реакционную смесь вводят амины.
Схема 3.38
Ограничения, накладываемые на формальную электрофильную компоненту реакции Соногаширы те же, что и в других реакциях кросс-сочетания. Реакция Соногаширы позволяет получать производные ацетиленов, труднодоступные другими методами (схема 3.39).
Схема 3.39
Реакция Хека
Реакция Хека представляет особый интерес для синтеза диенов и винилбензолов заданной конфигурации и состоит во взаимодействии винилгалогенидов или арилгалогенидов с алкенами в присутствии комплексов палладия (схема 3.40) [17, 18]. Высокая стереоселективность реакции Хека в отношении конфигурации образующихся алкенов обозначила интерес к ее изучению. Реакция Хека формально протекает как замещение атома водорода у атома углерода несущего наименее объемный заместитель с обращением конфигурации двойной связи (схема 3.40).
Схема 3.40 Это правило позволяет предсказывать стереохимический результат реакции. В частности, при проведении реакции Хека с участием монозамещенных алкенов всегда образуются продукты (Е)-конфигурации. Механизм реакции Хека включает стадии окислительного присоединения галогенида или трифлата, последующую координацию алкена, внедрение и син-элиминирование НРdХ. Каталитический цикл реакции Хека приведен на схеме 3.41.
Схема 3.41
Региоселективность реакции Хека контролируется пространственными факторами, так что замещение водорода происходит при наименее пространственно затрудненном тригональном углеродном центре. Стереоселективность реакции обеспечивается исключительным син-элиминированием НРdХ (схема 3.41). Реакция Хека протекает только с участием бромпроизводных, йодпроизводных и трифлатов винильных и арильных субстратов. В реакцию Хека вступают почти любые алкены, однако наилучшие результаты достигаются при использовании электронодефицитных алкенов (схема 3.42).
Схема 3.42
Реакция метатезиса
Реакция метатезиса позволяет проводить взаимные трансформации алкенов и состоит в воздействии на алкен или смесь алкенов соответствующего катализатора. Реакция метатезиса алкенов протекает согласно общей схеме, приведенной ниже:
Схема 3.43
Реакция метатезиса обратима и в общем случае приводит к смеси изомерных алкенов, поэтому основные усилия были направлены на оптимизацию условий проведения реакции и разработку каталитических систем, проявляющих высокую активность в мягких условиях. Первоначально реакцию метатезиса проводили в условиях термолиза алкенов над триоксидом вольфрама. Однако триоксид вольфрама совершенно не проявляет каталитической активности при температурах, близких к комнатной, что ограничивало область применения реакции метатезиса лишь простейшими алкенами. За последние полвека условия проведения реакции метатезиса и каталитические системы метатезиса претерпели драматические изменения. Если первоначально реакцию метатезиса проводили в условиях гетерогенного катализа при повышенных температурах, то в настоящее время она реализуется преимущественно в условиях гомогенного катализа при комнатной или даже пониженной температуре. В настоящее время разработано значительное число каталитических систем для проведения реакции метатезиса с участием алкенов самого разнообразного строения (схема 3.44) [19,20].
Схема 3.44
Катализатор 1-Мо, широкоизвестный как катализатор Шрока предложенный в 1990г. является одним из наиболее эффективных, однако ввиду его исключительной чувствительности к кислороду и влаге воздуха, работа с ним требует специального оборудования. В 1995г. был предложен катализатор 3-Ru, обладающий высокой каталитической активностью и не требующий исключения влаги воздуха при работе с ним. Катализатор 3-Ru широко известен как катализатор Граббса первого поколения и зарекомендовал себя как один из наиболее универсальных катализаторов метатезиса. Катализатор Граббса 3-Ru коммерчески доступен, его синтезируют по схеме, приведенной ниже:
Схема 3.45
В конце 1990-х гг. было показано, что замена одного трифенилфосфинового лиганда на имидазолиновый лиганд приводит к резкому увеличению активности катализатора. Катализаторы, содержащие имидазолиновый лиганд (например, 4-Ru), известны как катализаторы Граббса второго поколения. Последующие работы по оптимизации структуры катализаторов метатезиса состояли в вариациях лигандов катализаторов Граббса второго поколения 4-Ru. В частности было показано, что катализатор 5-Ru, в котором рутений хелатирован кислородом изопропоксигруппы имеет одновременно повышенную стабильность и может быть выделен хроматографически из системы по завершению реакции, а также обладает высокой каталитической активностью. Область применения реакции метатезиса включает синтез алкенов, диенов и алкинов самого различного строения, а также полимеризацию ненасыщенных мономеров. Классификация процессов метатезиса по их формальному результату позволяет выделить четыре типа реакции метатезиса алкенов [21]: 1. Кросс-метатезис (СМ):
2. Метатезис с образованием цикла (RCM):
3. Полимеризация с раскрытием цикла (ROMP):
4. Полимеризация при метатезисе ациклического диена (ADMET):
Несмотря на то, что реакция метатезиса в алифатическом ряду была открыта в 1964 г. Бэнксом и Бэйли, ее механизм длительное время оставался неясным. В настоящее время общепризнан механизм, предложенный в 1970 г. французскими химиками И. Шовеном и Ж. Л. Эриссоном [22], предполагающий образование металлциклобутановых интермедиатов и представленный на схеме 3.46.
Схема 3.46
Механизм реакции метатезиса включает взаимодействие карбена, координированного на атоме металла, с алкеном, приводя к образованию металлциклобутанового интермедиата. Металлциклобутановый интермедиат обратимо фрагментирует с потерей молекулы этилена, тем самым регенерируется карбеновый реакционный центр. Последующее взаимодействие с другим (или этим же) алкеном приводит к образованию второго металлциклобутанового интермедиата, который также фрагментирует, выделяя молекулу продукта и регенерируя катализатор. Пути создания карбенового центра, координированного по атому переходного металла, могут быть самыми различными. Первые гомогенные вольфрамовые катализаторы предполагали образование активных центров при взаимодействии гексахлорида вольфрама и диэтилалюминийхлорида или какого-либо другого эквивалента алкильного карбаниона. Такой путь приводит к удовлетворительным результатам, однако проведение реакции метатезиса было сопряжено с работой в токе инертного газа в отсутствии влаги и паров любых других протонных растворителей даже в следовых количествах. Формирование карбенового активного центра в системе гексахлорид вольфрама – диэтилалюминийхлорид представлено на схеме 3.47.
Схема 3.47
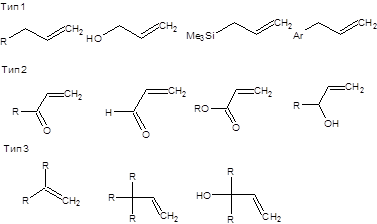
До разработки катализатора 1-Мо основное значение имели каталитические системы метатезиса на основе гексахлорида вольфрама, например WCl 6 - Et 2 AlCl, WCl 6 -Ме4 Sn, WCl 6 -Ме Li. Основным недостатком этих каталитических систем была относительно низкая активность и очень низкая селективность, поэтому принципиальным прорывом стало применение катализаторов, содержащих уже заранее готовый активный карбеновый центр, которые были приведены на схеме 3.96. Введение рутениевых катализаторов в синтетическую практику позволило проводить метатезис не только в присутствии кислорода воздуха и воды, но и в присутствии других функциональных групп, таких как спиртовая, альдегидная, кето-группа, сложноэфирная и даже карбоксильная. Амины отравляют рутениевые катализаторы, однако их метатезис наблюдается при их предварительном превращении в ацильные производные или аммонийные соли. Реакция кросс-метатезиса алкенов состоит во взаимодействии двух различных терминальных алкенов с образованием интернального алкена и этилена. Ранее приведенная схема кросс-метатезиса, кажется на первой взгляд весьма простой, однако в действительности ситуация несколько более сложна. Так при взаимодействии двух различных алкенов в условиях реакции метатезиса в общем случае может образовываться смесь трех различных алкенов (без учета геометрической изомерии), соответствующих гомодимеризации каждого из участвующих алкенов, а также требуемого продукта перекрестного взаимодействия. Ситуация дополнительно осложняется образованием различных геометрических изомеров получаемых алкенов. Преимущественное направление реакции определяется строением исходных алкенов и в меньшей степени типом используемого катализатора, хотя его влияние на региоселективность и стереоселективность реакции существенно. По своей способности к гомодимеризации все алкены можно разделить на три группы. К первой группе относятся алкены с незамещенным аллильным фрагментом по СН2-группе (терминальные алкены, первичные аллиловые спирты и их эфиры, аллиламины, защищенные по аминогруппе, первичные аллилсульфиды). Алкены первого типа легко гомодимеризуются в условиях реакции метатезиса. Ко второй группе относятся акролеин, акрилаты, винилкетоны, а также вторичные аллильные спирты и их эфиры. Алкены второй группы подвержены медленной гомодимеризации в условиях реакции метатезиса. К алкенам третьего типа относятся 1,1-дизамещенные терминальные алкены и алкены с полностью замещенным аллильным положением. Алкены третьего типа не гомодимеризуются, но вступают в реакцию кросс-метатезиса с алкенами двух других типов. Классификация алкенов по их способности к гомодимеризации в условиях реакции метатезиса приведена на схеме 3.48 [23].
Схема 3.49 Кросс-метатезис алкенов первого типа приводит к статистической смеси продуктов, с мольным отношением геометрических изомеровЕ/Z от 3 до 10. Кросс-метатезис алкенов первого типа может быть использован в препаративных целях, если один из участвующих алкенов имеет простую структуру и дешев, так как для успешного проведения реакции требуется не менее четырехкратного мольного избытка одного из алкенов. Перекрестный метатезис алкенов первого и второго (третьего) типов часто возможно провести успешно при использовании двукратного мольного избытка одного из алкенов или даже при их эквимолярном соотношении. Соотношение геометрических изомеров в этом случае смещено еще более в сторону образования наиболее термодинамически устойчивого (Е)-изомера. Взаимодействие алкенов второго и третьего типов осложнено возможностью побочной гомодимеризации алкенов второго типа. Если скорость перекрестного метатезиса алкенов превышает скорость их гомодимеризации, то продукт перекрестного метатезиса может быть получен. Низкая стереоселективность процесса перекрестного метатезиса алкенов второго и третьего типов ограничивает его синтетическое применение. Таким образом, реальное синтетическое значение кросс-метатезиса ограничено взаимодействием алкенов первого типа с алкенами второго и третьего типов. Примеры использования перекрестного метатезиса алкенов различных типов приведены на схеме 3.49.
Схема 3.49
|
|||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2021-05-27; просмотров: 446; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.145.108.9 (0.119 с.) |
|||||||||||||||||||||||||||