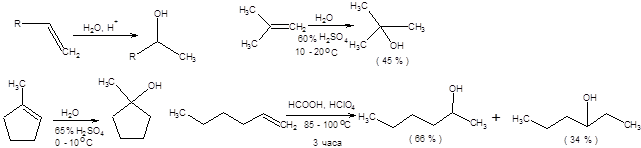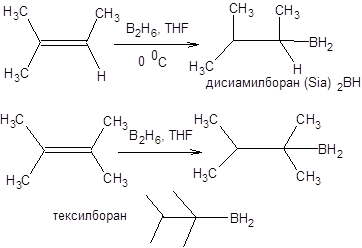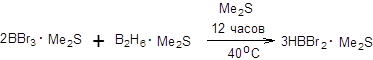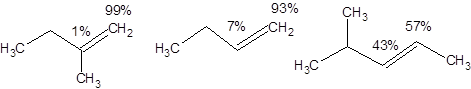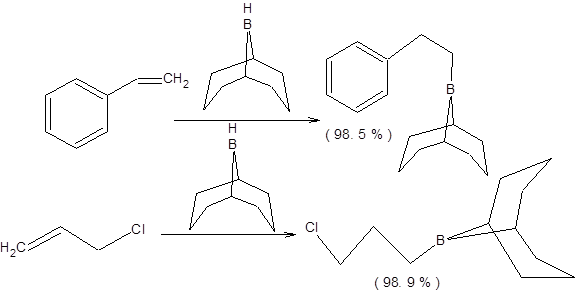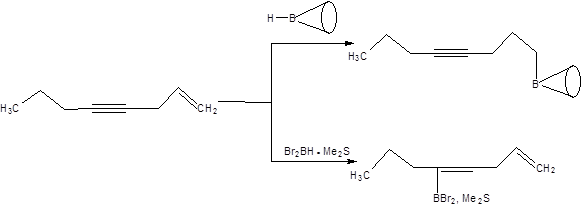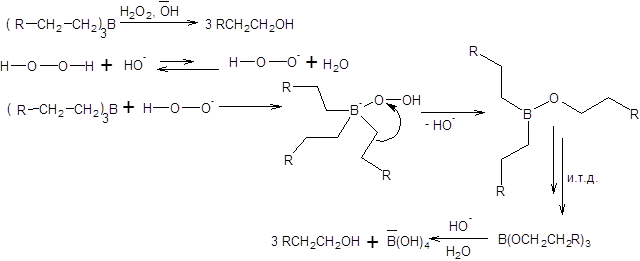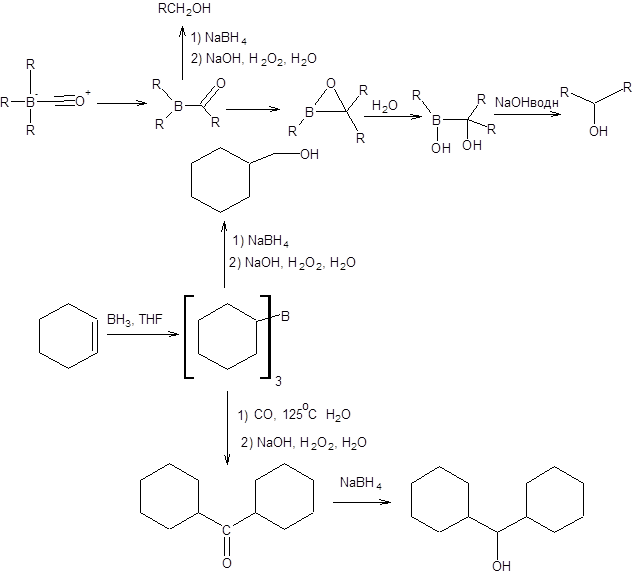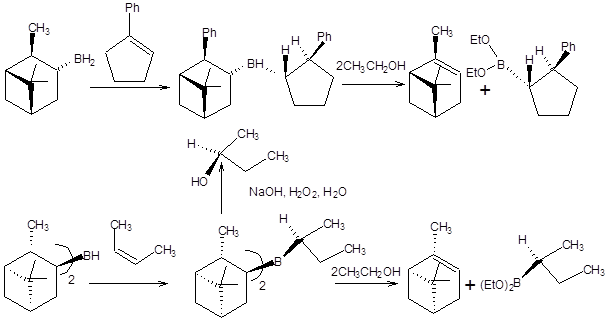Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Кислотно - катализируемая гидратация алкенов
Кислотно - катализируемая гидратация алкенов – один из исторически первых методов синтеза спиртов, однако его эффективность весьма мала, так как процесс осложняется перегруппировками типа Вагнера – Меервейна. При кислотно - катализируемой гидратации алкенов, преимущественно образуется продукт, отвечающий правилу Марковникова. В качестве катализатора кислотной природы используются минеральные кислоты, как правило, серная или фосфорная кислоты. Примеры использования кислотно-катализируемой гидратации алкенов представлены на схеме 4.38.
Схема 4.38
Удачной альтернативой методу кислотно-катализируемой гидратации является двухстадийный метод оксимеркурирования – демеркурирования. Реакция оксимеркурирования – демеркурирования гораздо более селективна и позволяет получать продукты марковниковской гидратации с высокими выходами. Механизм реакции оксимеркурирования – демеркурирования, а также некоторые примеры ее практического применения представлены на схеме 4.39.
Схема 4.39 Очевидные преимущества метода оксимеркурирования – демеркурирования перед кислотно-катализируемой гидратацией алкенов практически вытеснили последнюю реакцию из практики лабораторного органического синтеза. Вместе с тем кислотно-катализируемая гидратация этилена и пропилена остается промышленным методом получения этанола и изопропанола соответственно, находящих применение в качестве полярных протонных растворителей. Гидроборирование алкенов Гидроборирование алкенов приводит к образованию алкилборанов – важных промежуточных продуктов органического синтеза. Исходя из алкилборанов, возможно осуществить синтез: алканов, алкенов, галогенопроизводных, спиртов и кетонов. Из всего многообразия реакций, свойственных алкилборанам, в настоящем разделе рассматриваются только те, которые приводят к образованию спиртов. Гидроборирование алкенов состоит в присоединении диборана или других боранов по двойной углерод – углеродной связи, согласно схеме приведенной ниже [19, 20]:
Схема 4.40 Гидроборирование алкенов происходит региоселективно против правила Марковникова и контролируется преимущественно пространственными факторами. Гидроборирование алкенов происходит как син-присоединение по согласованному механизму (схема 4.41).
Схема 4.41 Гидроборирование пространственно незатрудненных алкенов не ограничивается присоединением одного эквивалента алкена, и продуктами реакции являются триалкилбораны. Иным образом дело обстоит при гидроборировании пространственно затрудненных алкенов, в этом случае возможно образование диалкилборанов и моноалкилборанов. Так эквивалент борана взаимодействует только с двумя эквивалентами 3-метилбутена-2 с образованием бис-(3-метил-2-бутил)-борана, известного как дисиамилборан, а тетраметилэтилен присоединяет всего один эквивалент борана, образуя тексилборан (схема 4.42).
Схема 4.42 Тексилборан и дисиамилборан способны гидроборировать пространственно незатрудненные алкены, кроме того, реакции с участием этих реагентов протекают более селективно, чем при использовании диборана в качестве гидроборирующего реагента. Также широкое распространение в синтетической практике приобрел 9-борабицикло[3,3,1]нонан (9-BBN), который получают гидроборированием 1,5-циклооктадиена (схема 4.43).
Схема 4.43 К числу весьма полезных гидроборирующих агентов относится дибромборан, получаемый взаимодействием трибромида бора и диборана в среде диметилсульфида в требуемых стехиометрических отношениях (схема 4.44).
Схема 4.44
Использование диборана в качестве селективного гидроборирующего реагента встречает трудности, если разница в объеме заместителей при двойной связи невелика. Так по мере уменьшения разницы в объемах заместителей при двойной связи региоселективность гидроборирования существенно снижается, как это показано на схеме 4.45.
Схема 4.45
Для увеличения селективности гидроборирования эффективно применяют рассмотренные выше пространственно затрудненные гидроборирующие реагенты, причем с ростом пространственных требований борана возрастает селективность гидроборирования по наименее пространственно затрудненному атому углерода двойной связи исходного алкена. Дисиамилборан – реагент с высокими пространственными требованиями позволяет селективно гидроборировать интернальные алкены и крайне чувствителен к объему заместителей при двойной связи. Дисиамилборан также обладает высокой региоселективностью при гидроборировании терминальных алкенов (схема 4.46).
Схема 4.46
Еще большей селективностью обладает 9-BBN, который предъявляет еще более высокие пространственные требования, чем дисиамилборан и обеспечивает, почти количественный выход боранов в последних двух рассмотренных реакциях (схема 4.46). Селективное гидроборирование стирола и аллилхлорида составляет известную трудность, так в случае применения диборана выходы продуктов антимарковниковского присоединения составляют соответственно 80 % и 60 %. Применение 9-BBN в качестве гидроборирующего реагента позволяет провести гидроборирование этих субстратов с большей селективностью против правила Марковникова (схема 4.47).
Схема 4.47
Тексилборан применяется для селективного гидроборирования (Z)-двойной связи в присутствии (Е)-двойной связи, ввиду того, что (Е)-алкены гидроборируются в 100 раз медленнее, чем алкены (Z)-конфигурации. Особый интерес представляет дибромборан, применяемый в виде комплекса с диметилсульфидом. Проводя сравнение региоселективности гидроборирования алкенов дибромбораном и дисиамилбораном можно заключить, что дисиамилборан гидроборирует наименее замещенную двойную связь, тогда как дибромборан наиболее замещенную, причем оба реагента присоединяются с высокой селективностью против правила Марковникова (схема 4.48).
Схема 4.48
Дибромборан преимущественно гидроборирует тройную связь в присутствии двойной связи, тогда как 9-BBN отличается поразительной инертностью по отношению к тройной связи и селективно гидроборирует двойную связь в присутствии тройной (схема 4.49).
Схема 4.49
Возможность проведения хемоселективного и региоселективного гидроборирования кратных связей открывает широкие синтетические возможности, в том числе и для получения спиртов различного строения. Алкилбораны превращаются в соответствующие спирты под действием щелочного раствора пероксида водорода, причем реакция протекает гладко, с высоким выходом спирта. Общая схема и механизм расщепления триалкилборанов щелочным раствором перекиси водорода приведены ниже:
Схема 4.50
Альтернативный метод получения спиртов, исходя из триалкилборанов, основан на их предварительном взаимодействии с монооксидом углерода. Получаемый аддукт далее может быть превращен в первичные, вторичные и третичные спирты. Третичные спирты легко образуются согласно схеме, представленной ниже:
Схема 4.51
Синтез первичных и вторичных спиртов из аддукта окиси углерода и триалкилборанов осуществляется введением реагентов, препятствующих дальнейшей миграции алкильных групп от атома бора к углероду. Первичные спирты образуются путем введения в реакционную систему боргидрида натрия, который восстанавливает интермедиат после миграции только одной алкильной группы, тем самым препятствуя миграции двух остальных. Введение в реакционную систему воды останавливает миграцию последней группы, последующий щелочной гидролиз и восстановление приводят к образованию вторичного спирта (схема 4.52).
Схема 4.52
Рассмотренным способом возможно получение лишь симметричных вторичных спиртов, что составляет существенное ограничение метода. Это ограничение можно устранить, если одна из групп исходного триалкилборана будет пространственно затрудненной, например, тексильной, а ее миграция будет невозможна или будет происходить с малой скоростью. Введение хиральных боранов в синтетическую практику позволило проводить асимметрический синтез спиртов и бороновых кислот. В качестве хиральных боранов наиболее часто используются монопинанилборан и дипинанилборан (Схема 4.53) [21].
Схема 4.53
Список литературы
1. Grignard V. C. R. // Acad. Sci. 1900. V. 130. P.1322. 2. Toda N., Ori M., Takami K., Tago K., Kogen H. // Org. Lett. 2003.V. 5.P. 269. 3. Imamoto T., Sugiura Y., Takiyama N. // Tetrahedron Lett. 1984.V. 25.P. 4233. 4. Imamoto T., Kusumoto T., Tawarayama Y., Sugiura Y., MitaT., HatanakaY., Yokoyama M. // J. Org. Chem. 1984. V. 49. P. 3904. 5. Реутов О. А., Курц А. Л., Бутин К. П. Органическая химия. М.: Бином. Лаборатория знаний. 2004. Т. 2. С. 79. 6. Luche J. L. // J. Am. Chem. Soc. 1978. V. 100. P. 2226. 7. Cram D. J., Elhafez F. A. A. // J. Am. Chem. Soc. 1952.V.74.P. 5828. 8. Смит В. А., Дильман А. Д. Основы современного органического синтеза. М.: Бином. Лаборатория знаний, 2009. C. 148. 9. Anh N. T. // Top. Curr. Chem. 1980. V. 88. P. 145. 10. Midland M. M. // Tetrahedron. 1984 V. 40. P. 1371. 11. Brown H. C., Chandrasekharan J., Ramachandran P. V. // J. Am. Chem. Soc. 1988. V. 110. P. 1539. 12. Corey E. J., Bakshi R. K., Shibаtа S. // J. Am. Chem. Soc. 1987. V.109. Р. 5551. 13. Itsuno S. // Org. React. 1998. V. 52. P. 395. 14. Corey E. J., Link J. O. // J. Am. Chem. Soc.1992.V. 114. P. 1906. 15. Noyori R. Asymmetric catalysis in organic synthesis. New York. Wiley, 1994. 378 Р. 16. Noyori R. // Angew. Chem. Int. Ed. 2002. V. 41. P. 2008. 17. Bouveault L., Blanc G. // Compt. Rend. 1903. V. 136. P. 1676. 18. Aremo N., Hase T. // Org. React. 2001. V. 42. P. 3637. 19. Нодзаки Х. Современные направления в органическом синтезе. М.: Мир, 1986. С. 339 – 361. 20. Brewster J. H., Negishi E. // Science. 1980. V. 207. P. 44. 21. Brown H. C., Jadhav P. K., Desai M. C. // J. Am. Chem. Soc. 1982. V.104. P. 4303.
Лекция 5. Синтез аминов
|
|||||||
|
|
Последнее изменение этой страницы: 2021-05-27; просмотров: 461; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.216.186.164 (0.034 с.) |