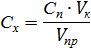Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Определение компонентов технологических растворов, природных и сточных вод
Методом потенциометрического титрования можно определять различные компоненты технологических растворов, сточных и природных вод на основе реакций кислотно-основного и окислительно-восстановительного взаимодействия, реакций осаждения и комплексообразования. В таблице 2.6 даны примеры компонентов технологических растворов, природных и сточных вод, которые могут быть даны студенту для определений при выполнении учебно-исследовательской работы. По таблице выбирают титрант и индикаторный электрод, которые будут использованы при проведении потенциометрического титрования.
Порядок выполнения работ 1. Подготовка к работе рН-метра и электродов. 2. Выбор титранта и установление концентрации титранта методом потенциометрического титрования стандартного раствора. Титрование повторяют 2-3 раза. Результаты стандартизации титранта записываются в таблицу 2.7 и рассчитывают среднее значение концентрации титранта, моль/л. 3. Расчет объема пробы анализируемого раствора и разбавление пробы в мерной колбе (при необходимости). 4. Потенциометрическое титрование анализируемого раствора. Первое титрование (ориентировочное) проводят, прибавляя по 0,40 мл титранта. На кривой титрования выделяют область, где наблюдается скачок потенциала. Точное титрование повторяют не менее трех раз, прибавляя в области ожидаемого скачка потенциала по 0,20 мл титранта. Результаты титрования записывают по форме, приведенной в таблице 2.8. После каждого титрования строят кривую титрования и определяют объем титранта в КТТ, мл. Все кривые титрования строят на одном листе бумаги, каждый раз сдвигая начало координат вправо на 2-4 см.
Таблица 2.6 - Примеры компонентов технологических растворов
5. Расчет содержания определяемого компонента Х в растворе по результатам каждого из 3-4 титрований проводят по формуле С (f эквХ) = С (f эквТ). V КТТ/ V пр, моль/л. Рассчитанную концентрацию по заданию УИР выражают в г/л (мг/л, %). 6. Математическая обработка результатов параллельных определений концентрации компонента в анализируемом растворе проводится с целью оценки правильности и воспроизводимости (см. раздел 1.3.). Обработанные данные представляют в виде таблицы 1.1.
Таблица 2. 7 – Установление концентрации титранта (название или формула)
Таблица 2.8 – Результаты определения (название или формула)
Ниже приведены примеры выполнения определения компонентов различных растворов методом потенциометрического титрования.
Работа 1. Определение содержания НС1 в растворе
Цель работы: Определение содержания соляной кислоты в растворе методом потенциометрического титрования раствором NaOH. Оценка правильности и воспроизводимости результатов определения.
Определение содержания НС1 в растворе при титровании основано на реакции кислотно-основного взаимодействия:
HС1 + NaOH = NaС1 + Н2О.
Реактивы, посуда, приборы Гидроксид натрия NaOH - 0,1 М раствор; Раствор серной кислоты, С (1/2H2SO4) = 0,1000 М; Установка для потенциометрического титрования; Индикаторный электрод – стеклянный;
Электрод сравнения – хлоридсеребряный; Пипетки вместимостью 5,00 и 10,00 мл.
Выполнение работы 1. Установление концентрации титранта. Концентрацию гидроксида натрия устанавливают при потенциометрическом титрования 5,00 мл стандартного раствора серной кислоты, С (1/2H2SO4) = 0,1000 М, раствором NaOH, руководствуясь положениями раздела 2.2.1. 2. Определение содержания кислоты в растворе. В стаканчик для титрованиямерной пипеткой переносят 10,00 мл анализируемого раствора кислоты НС1. Опускают в раствор размешиватель, индикаторный электрод и электрод сравнения, доливают дистиллированной воды, чтобы нижняя часть электродов была погружена в раствор. Включают мешалку. Заполняют бюретку стандартным раствором титранта NaOH. Уровень раствора автоматически устанавливается на нулевом делении. Проводят потенциометрическое титрование, строят кривую титрования, по которой находят КТТ и V КТТ. Точное титрование повторяют не менее трех раз, прибавляя в области ожидаемого скачка потенциала по 0,20 мл титранта. Результаты титрования записывают по форме, приведенной в таблице 2.8. По результатам титрования вычисляют концентрацию НС1 в растворе, моль/л и выражают в г/л и %. Все результаты представляют в виде таблиц 2.7 – 2.8. Проводят математическую обработку результатов параллельных определений концентрации компонента С, г/л. Обработанные данные представляют в виде таблицы 1.1 (см. раздел 1.3).
Работа 2. Определение содержания хлорида натрия в растворе
Цель работы: Определение содержания хлорида натрия в растворе методом потенциометрического титрования раствором нитрата серебра. Оценка правильности и воспроизводимости результатов определения.
Определение содержания NaС1 в растворе при титровании основано на реакции осаждения NaС1 + AgNO3 = AgС1↓ + NaNO3.
Реактивы, посуда, приборы Нитрат серебра AgNO3 – 0,1 М раствор; Установка для потенциометрического титрования; Индикаторный электрод – серебряный; Электрод сравнения – хлоридсеребряный; Пипетки вместимостью 5,00 мл и 10,00мл.
Выполнение работы 1. Установление концентрации титранта. Концентрацию раствора AgNO3 предварительно устанавливают путем титрования 5,00 мл стандартного раствора хлорида калия С (КС1) = 0,1000 М, следуя рекомендациям п.2.2.1. 2. Определение содержания кислоты в растворе. В стаканчик для титрованиямерной пипеткой переносят 10,00 мл анализируемого раствора хлорида натрия. Опускают в раствор размешиватель, серебряный и хлоридсеребряный электроды, доливают дистиллированной воды, чтобы нижняя часть электродов была погружена в раствор. Включают мешалку. Заполняют бюретку титрантом AgNO3. Уровень раствора автоматически устанавливается на нулевом делении. В соответствие с рекомендациями п.2.2.1 проводят потенциометрическое титрование, строят кривую титрования для нахождения КТТ и V КТТ. Проводят 3-4 параллельных определений. Содержание NaС1 в растворе вычисляют по формуле
С (NaС1) = С (AgNO3). V КТТ/ V NaС1, моль/л.
Полученную концентрацию выражают в г/л и %. Все результаты представляют в виде таблиц 2.7 – 2.8. Проводят математическую обработку результатов параллельных определений концентрации компонента С, г/л. Обработанные данные представляют в виде таблицы 1.1 (см. раздел 1.3).
Работа 3. Определение Н2О2 в растворе Цель работы: Определение содержания пероксида водорода в отбеливающем растворе методом потенциометрического титрования раствором перманганата калия. Оценка правильности и воспроизводимости результатов определения.
Определение содержания Н2О2 в отбеливающих растворах основано на реакции с перманганатом калия, протекающей в кислой среде
5H2O2 + 2KMnO4 + 3H2SO4 = 2MnSO4 + 5O2 + K2SO4 + 8H2O.
Реактивы, посуда, приборы Стандартный раствор перманганата калия, С (1/5 KMnO4) = 0,1000 М; Раствор серной кислоты (1:4); Пипетка вместимостью 5,00 мл; Бюретка вместимостью 25,00 мл; Иономер (рН-метр) любого типа; Индикаторный электрод – платиновый; Электрод сравнения – хлоридсеребряный; Стаканчик для титрования, размешиватель; Магнитная мешалка.
Выполнение работы В работе используют стандартный раствор перманганата калия (значение концентрации выражено четырьмя значащими цифрами), поэтому его концентрацию устанавливать не следует. 1. Расчет объема пробы анализируемого раствора. Зная ориентировочное содержание пероксида водорода в анализируемом растворе, концентрацию титранта по закону эквивалентности рассчитывают объем пробы, который надо взять, чтобы на титрование пошло не более половины объема титранта, находящегося в бюретке. Выбирают пипетку для отбора пробы, вместимость которой близка к рассчитанному значению. 2. Определение содержания Н2О2 в анализируемом растворе Заполняют бюретку титрантом, подключают электроды к прибору на задней стенке корпуса, включают прибор в сеть (тумблером на приборе) и прогревают в течение 20 минут. В стаканчик для титрования выбранной пипеткой переносят аликвоту (точный объем) анализируемого раствора, добавляют 5 мл раствора H2SO4 (1:4), опускают размешиватель, электроды (индикаторный и электрод сравнения). При необходимости доливают в стаканчик дистиллированной воды так, чтобы электроды были погружены в раствор на 1,5 – 2 см. Включают мешалку. Размешиватель при вращении не должен задевать электроды. Перемешивание осуществляют в течение всего титрования. Проводят титрование, строят кривую титрования для нахождения КТТ и V КТТ. Для набора статистических данных повторяют определение 4-5 раз. По результатам каждого титрования строят кривую титрования и определяют объем титранта в КТТ. Рассчитывают молярную концентрацию эквивалента H2O2 С (1/2Н2О2), моль/л и массовую концентрацию, г/л.
3. Математическую обработку результатов проводят по значениям концентраций, г/л. Все результаты представляют в виде таблиц 2.8 и 1.1.
Работа 4. Определение дихромата калия в растворе Цель работы: Определение содержания дихромата калия в ванне хромирования методом потенциометрического титрования. Оценка правильности и воспроизводимости результатов определения. При титровании дихромата калия солью Мора в кислой среде протекает окислительно-восстановительная реакция
6FeSO4 + K2Cr2O7 + 7H2SO4 = Cr2(SO4)3 + K2SO4 + 3Fe2(SO4)3 + 7H2O.
Конечная точка титрования (КТТ) устанавливают потенциометрически.
Реактивы, посуда, приборы Сухая соль K2Cr2O7, ч.д.а. или раствор С(1/6 K2Cr2O7) = 0,1000 моль/л; Соль Мора, раствор С(FeSO4) ~ 0,1 моль/л; Раствор серной кислоты - 1 моль/л; Колбы мерные вместимостью 100,0 мл; Пипетка вместимостью 10,00 мл; Бюретка вместимостью 25,00 мл; Иономер (рН-метр) любого типа; Индикаторный электрод – платиновый; Электрод сравнения – хлоридсеребряный; Стаканчик для титрования, размешиватель; Магнитная мешалка.
Порядок выполнения работы
1. Приготовление раствора установочного вещества дихромата калия. 2. Установление концентрации раствора соли Мора. 3. Определение содержания дихромата калия в анализируемом растворе. 4. Математическая обработка результатов.
Выполнение работы 1. Приготовление раствора установочного вещества дихромата калия. Для установления концентрации соли Мора используют стандартный раствор дихромата калия. Установочный раствор дихромата калия готовят по точной навеске соли K2Cr2O7 в мерной колбе. Навеску соли, необходимую для приготовления 100 мл раствора с С(1/6K2Cr2O7) ~ 0,1 моль/л, рассчитывают по формуле
Рассчитанную массу соли взвешивают первоначально на технических, а затем на аналитических весах и переносят в мерную колбу вместимостью 100,0 мл, доливают до метки дистиллированной водой и раствор хорошо перемешивают. Концентрацию полученного раствора рассчитывают по формуле
где m – масса навески, полученная после взвешивания на аналитических весах, V к – объем колбы. При наличии можно использовать готовый стандартный раствор С(1/6 K2Cr2O7) = 0,1000 моль/л 2. Установление концентрации раствора соли Мора. Установление концентрации раствора соли Мора по дихромату калия проводят путем потенциометрического титрования в кислой среде. Бюретку заполняют раствором соли Мора. В стаканчик для титрования переносят пипеткой 5,00 мл стандартного раствора K2Cr2O7, добавляют 10 мл 1 М раствора H2SO4 и 10-15 мл дистиллированной воды. В стаканчик опускают размешиватель, погружают в раствор электроды. Включают мешалку и начинают титрование, добавляя по 0,4 мл, а вблизи точки эквивалентности по 0,2 мл. После прибавления каждой порции титранта величину потенциала записывают по форме, приведенной в таблице 2.5. Титрование прекращают, когда после скачка потенциала прибавление очередной порции титранта почти не будет вызывать изменение потенциала. Титрование повторяют два-три раза.
После окончания титрования выключают мешалку, промывают электроды дистиллированной водой. По полученным данным строят кривые титрования Е = f (V титр), по которым определяют объем в конечной точке титрования V КТТ. Рассчитывают концентрацию соли Мора по закону эквивалентности. Результаты оформляют в виде таблицы 2.7. 4. Определение содержания дихромата калия в ванне хромирования. Рассчитывают ~ объем пробы анализируемого раствора для титрования, исходя из предположительного состава ванны хромирования, который указывается в задании на УИР. Для отбора пробы выбирают пипетку, вместимость которой близка к рассчитанному значению (см.п.2.2.2). В стаканчик для титрования выбранной пипеткой переносят аликвоту (точный объем) анализируемого раствора, добавляют 10 мл 1М раствора H2SO4, опускают размешиватель, электроды. При необходимости доливают в стаканчик дистиллированной воды так, чтобы электроды были погружены в раствор на 1,5 – 2 см. Включают мешалку. Размешиватель при вращении не должен задевать электроды. Перемешивание осуществляют в течение всего титрования. Проводят ориентировочное титрование раствором соли Мора, добавляя по 0,4 мл титранта. Результаты измерений потенциала записывают в виде таблицы 2.5. По полученным данным строят кривую титрования для нахождения КТТ и V КТТ. Титрование повторяют не менее трех раз, добавляя в области скачка потенциала по 0,2 мл титранта. Концентрацию дихромата калия в анализируемом растворе рассчитывают по закону эквивалентности. Результаты определения записывают в виде таблицы 2.8. 5. Проводят математическую обработку результатов определения концентрации K2Cr2O7 в анализируемом растворе (см. раздел 1.3.) и данные представляют в виде таблицы 1.1.
2.3. Контрольные вопросы 1. На чем основаны методы потенциометрического титрования и прямой потенциометрии? 2. Напишите выражение зависимости потенциала ионоселективного электрода от активности (концентрации) ионоселективного электрода. 3. Назовите индикаторный электроды и электрод сравнения, которые используются в методах потенциометрического титрования и ионометрии. 4. Назовите титрант, индикаторный электрод и электрод сравнения для определения хлорид-ионов в растворе. 5. Назовите индикаторный электрод и титрант для определения кислот; оснований. 6. Назовите индикаторный электрод для определения соединений с окислительно-восстановительными свойствами. 7. Напишите реакции, которые используются при определении кислоты НС1 по катиону и по аниону методом потенциометрического титрования. 8. От каких факторов зависит выбор индикаторного электрода? 9. Какие требования предъявляют к электроду сравнения? 10. С какой целью, и в каких координатах строят кривую титрования? 11. Как определяют КТТ методом потенциометрического титрования? 12. Какой закон лежит в основе расчетов при титровании? Запишите его выражение. 13. Перечислите основные узлы установки для потенциометрического титрования и прямой потенциометрии и их назначение. 14. Назовите основные достоинства методов потенциометрического титрования и ионометрии. 15. В каких координатах строят график в ионометрии? 16. С какой целью снимают градуировочный график? 17. Приведите последовательность определения ионов в растворе методом ионометрии с использованием градуировочного графика. 18. Какой электрод служит для измерения рН раствора? 19. Вычислите –lg0,0001. 20. Как изменится рН раствора, если молярная концентрация раствора НС1 уменьшилась в 10 раз?
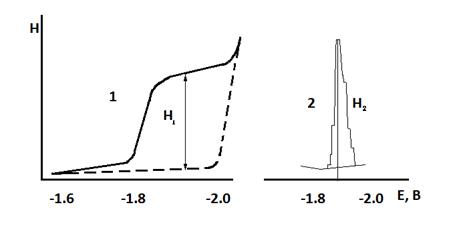
3. МЕТОДЫ ПОЛЯРОГРАФИИ Полярография является разделом вольтамперометрии, где рабочим электродом служит ртутный капающий электрод (РКЭ). Она основана на изучении вольтамперных (поляризационных) кривых, представляющих собой зависимость силы тока от развертки внешнего напряжения при электролизе определяемого соединения. Основными условиями выполнения полярографического определения являются выбор электродов, фонового электролита (фона) и растворителя. Кроме того, определяемое соединение должно быть электроактивно. Электролиз проводится с использованием легко поляризуемого электрода с малой поверхностью, например, РКЭ, на котором и происходит электровосстановление или электроокисление вещества. В качестве вспомогательного электрода используют донную ртуть. Площадь поверхности ртутной капли в тысячи раз меньше поверхности донной ртути, поэтому налагаемое напряжение поляризует микроэлектрод. Концентрация фонового электролита превышает концентрацию определяемого соединения приблизительно в 1000 раз, что уменьшает сопротивление раствора, а также создает условия, при которых ионы определяемого вещества не участвуют в переносе заряда, и устраняет вероятность появления тока миграции. Перенос определяемых ионов к поверхности рабочего электрода осуществляется благодаря диффузии. Растворитель не должен окисляться или восстанавливаться в рабочей области потенциалов, где восстанавливается или окисляется определяемое вещество. В постояннотоковой (классической) полярографии полярограммы, представляют собой зависимость силы тока I от приложенного напряжения Е и записываются самописцем прибора в виде полярографической волны с высотой Н, мм, пропорциональной силе диффузионного тока (рисунок 3.1, кривая 1). В переменнотоковой полярографии на электроды подается не только линейно изменяющееся напряжение постоянного тока, но и переменное напряжение (синусоидальной, квадратноволновой или трапецеидальной формы) постоянной величины и небольшой амплитуды Полярограф регистрирует зависимость переменной составляющей тока от линейно изменяющегося напряжения постоянного тока. В этом случае полярограмма имеет вид пика (рисунок 3.1, кривая 2). Качественный полярографический анализ основан на измерении потенциала полуволны Е 1/2 (рисунок 3.1, кривая 1) или потенциала пика Е р (рисунок 1, кривая 2). При данных электродах, фоновом электролите и растворителе Е 1/2 и Е р не зависят от количества определяемого соединения, а зависят от его природы. В справочных таблицах по значениям потенциалов полуволны можно идентифицировать определяемые соединения. Количественный полярографический анализ основан на измерении силы предельного диффузионного тока Id в постояннотоковой полярографии или силы пикового тока Ip в переменнотоковой полярографии. Величины Id и Ip связаны с концентрацией определяемого соединения Сх линейными зависимостями Id = k Cx и Ip = k1 Cx.
На полярограммах обычно измеряют высоту волны или пика, которые также линейно зависят от концентрации определяемого соединения:
Hd = k ꞌ Cx; Hp = k ꞌ 1 Cx.
Рисунок 3.1 – Полярограммы в методах постояннотоковой (1) и переменнотоковой (2) полярографии.
3.1. Установка для полярографических определений
Принципиальная схема постояннотокового полярографа с ртутным капающим электродом представлена на рисунке 3.2. Установка состоит из ячейки (электролизёра) с ртутным капающим электродом и донной ртутью в качестве вспомогательного электрода, источника постоянного тока, делителя напряжения (реохорда), вольтметра и микроамперметра. Работу проводят в постояннотоковом режиме, вольтамперную кривую записывают на самописце. Кислород из полярографируемого раствора удаляют продуванием инертного газа. В полярографе предусмотрены различные режимы регистрации полярограмм: непрерывный, непрерывный с демпфированием (сглаживанием осцилляций) и таст – режим, при котором регистрация тока производится в определенный и очень короткий момент «жизни» капли. Представителем современных многофункциональных комплексов для проведения количественного и качественного анализа, различных электрохимических исследований является вольтамперометрический анализатор (полярограф) «Экотест - ВА», внешний вид которого представлен на рисунке 3.3.Полярограф сочетает в себе самые последние достижения вольтамперометрии и полярографии с сервисными возможностями компьютерного управления экспериментом, обработкой и протоколированием данных. Универсальный комплекс позволяет определять микроколичества тяжелых металлов, йода, селена и мышьяка, токсичных органических и неорганических компонентов в самых различных объектах методами инверсионной вольтамперометрии и полярографии. Анализатор «Экотест-ВА» обладает рядом эксплуатационных особенностей: · возможность определения нескольких элементов (марганец, цинк, кадмий, свинец, медь, висмут) одновременно из одной пробы; · высокая чувствительность, например, по Cd и Pb до 0,01 мкг/л; · низкая себестоимость единичного элементоопределения; · высокая производительность труда (особенно в роботизированном варианте); · отсутствие необходимости применения жидкой ртути; · работа без инертного газа; · автоматические режимы выполнения измерений и расчета концентраций; · работа по записанным в память методикам и количественная обработка результатов в автоматическом режиме; · возможность программирования анализатора для проведения до 100 измерений с различными параметрами.
Рисунок 3.2 – Схема полярографической установки: 1 – ячейка, 2 – сосуд с ртутью, 3 – микроамперметр, 4 – подвижный контакт, 5 – реохорд, 6 – источник постоянного тока, 7 – вольтметр Рисунок 3.3 – Внешний вид анализатора «Экотест - ВА»
«Экотест-ВА» является реальной экономичной альтернативой дорогостоящему атомно-абсорбционному методу анализа при определении микроколичеств тяжелых металлов, йода, селена и мышьяка.
Методика выполнения анализа
Установку к работе готовит инженер. Он выбирает из памяти компьютера необходимую программу определения. При необходимости изменяет начальные параметры снятия вольтамперных кривых: начальный и конечный потенциал развёртки, скорость развёртки потенциала, время накопления компонента на электроде и время его растворения, параметры очистки электрода и другие. Студент готовит серию стандартных растворов определяемого компонента для проверки линейности градуировочного графика, заливает в ячейку фоновый электролит и прописывает для проверки его чистоты. Затем прописывает стандартные растворы и строит градуировочный график. В случае его линейности прописывают анализируемый раствор и рассчитывают концентрацию определяемого компонента. Для количественного определения компонентов анализируемого раствора в полярографии применяют метод градуировочного графика и метод добавок, которые рассмотрены в разделе 1.1. При использовании метода градуировочного графика приготовление серии стандартных растворов путем разбавления исходного стандартного раствора проводят двумя способами: 1) непосредственно в полярографической ячейке, 2) в мерных колбах. В первом случае разбавление проводят в ячейке. Сначала в ячейку пипеткой переносят известное количество фонового электролита и снимают полярограмму. Затем пипеткой вместимостью 1,00 или 0,10 мл или калиброванным хроматографическим микрошприцом вместимостью 1,00 или 10,00 мкл вводят в фоновый электролит нужный объём стандартного раствора и снимают полярограмму. Затем ещё несколько раз вводят тот же объём стандартного раствора, каждый раз при этом снимая полярограммы. На полярограммах измеряют высоту полярографических волн или пиков и строят градуировочный график, предварительно рассчитав концентрации определяемого компонента в каждом растворе по формуле
где C ст и Ci – концентрации определяемого компонента в стандартном и полярографируемом растворах в моль/л, г/л, г/мл; V ст и V ф - объёмы добавленного стандартного раствора и фонового электролита в ячейке, мл. Во втором случае серию стандартных растворов готовят в мерных колбах. Для этого в пять колб пипеткой вносят от 1 до 5 необходимых объёмов стандартного раствора. Растворы доводят до метки фоновым электролитом. Приготовленные растворы последовательно переносят в полярографическую ячейку и снимают полярограммы. Объём раствора в ячейке не имеет значения, а концентрацию компонента в растворе рассчитывают по формуле
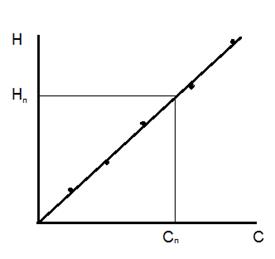
где C ст и Ci – концентрации определяемого компонента в стандартном иполярографируемом растворах в моль/л, г/л, г/мл; V ст и V к – объёмы стандартного раствора и мерной колбы. Значения Ci стандартных растворов и соответствующие им значения Hi заносят в таблицу и строят градуировочный график в координатах H – C. Порядок построения градуировочного графика приведен в п.1.1. График в определенном интервале концентраций представляет собой прямую, выходящую из начала координат, если выполняется зависимость Id = k Cx (рисунок 3.4). Линейность градуировочного графика следует периодически проверять, а при замене каких-либо реагентов, растворов, приборов, условий проведения анализа обязательно строят новую.
Рисунок 3.4 – Градуировочный график в полярографии
Рассчитывают пробу анализируемого раствора V пр таким образом, чтобы измеряемое значение H п в полярографируемом растворе не выходило за пределы градуировочной зависимости. Разбавление пробы фоновым электролитом также проводят непосредственно в полярографической ячейке или в мерной колбе. Если анализируемый раствор разбавляют непосредственно в полярографической ячейке, то пробу этого раствора пипеткой или микрошприцом переносят в полярографическую ячейку с известным объёмом фонового электролита и снимают полярограмму. По измеренному значению H п с помощью градуировочного графика (рисунок 4.4) или по уравнению прямой находят концентрацию определяемого компонента в полярографируемом растворе. Для уравнения градуировочного графика предварительно вычисляют коэффициенты а и в. Концентрацию определяемого компонента в анализируемом растворе рассчитывают по формуле
где Cx и C п – концентрации определяемого компонента в анализируемом и полярографируемом растворах, моль/л, г/л, г/мл; V ф и V пр – объёмы фонового электролита и пробы анализируемого раствора в ячейке, мл. Если анализируемый раствор разбавляют в мерной колбе, то рассчитанную пробу раствора пипеткой или микрошприцом переносят в мерную колбу и доводят до метки фоновым электролитом. Полученный раствор полярографируют. По измеренному значению H п с помощью градуировочного графика или по уравнению градуировочного графика находят концентрацию определяемого компонента в полярографируемом растворе. Далее рассчитывают концентрацию компонента в анализируемом растворе по формуле
где Cx и Сп - концентрации определяемого компонента в анализируемом и полярографируемом растворе, моль/л, г/л, г/мл; V к и V пр – объём мерной колбы и объём пробы анализируемого раствора, мл.
3.3. Исследование условий определения полярографическим методом
При определении компонентов технологических растворов, сточных и природных вод необходимо провести предварительные исследования, которые заключаются в установлении границ линейности градуировочного графика, проверке его стабильности во времени и установлении степени разбавления пробы. При установлении степени разбавления пробы нужно учитывать, что полярографический метод анализа используется для определения малых концентраций вещества. В постояннотоковой полярографии 10–5 – 10–3 моль/л. Обязательным является исследование влияния компонентов технологического раствора, природных или сточных вод на градуировочный график. Для этого при снятии полярограмм в фоновый электролит вводят не только стандартный раствор определяемого соединения, но и раствор компонентов, содержащихся в технологическом растворе, природной или сточной воде. Эти исследования позволяют выбрать метод количественного определения. Если градуировочная зависимость стабильна во времени, целесообразно использовать метод градуировочного графика. В противном случае, когда градуировочная зависимость не стабильна во времени или компоненты технологического раствора оказывают влияние на угол наклона графика, но при этом он выходит из начала координат, следует использовать метод добавок. В исследование условий определения обязательно должна входить проверка правильности полученных результатов, которую можно провести путём анализа модельного раствора с известной концентрацией определяемого компонента или сопоставлением результатов, полученных двумя независимыми методами.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2021-03-09; просмотров: 332; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.193.129 (0.183 с.) |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 .
.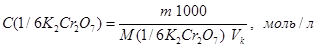 ,
,


 ,
, ,
,
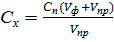 ,
,