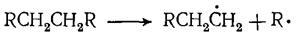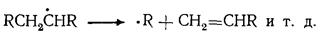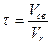Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Тема: алкилирование бензола этиленом (пропиленом)Стр 1 из 14Следующая ⇒
Лабораторная работа №1 Цель работы Изучение влияния мольного отношения поглощенного олефина к бензолу на состав реакционной массы и селективность по моноалкилбензолу при алкилировании бензола этиленом (пропиленом) в присутствии катализаторного комплекса хлорида алюминия. Методика выполнения работы
Реакторы Азот (из баллона) Бензол 97,5 г Этилен или пропилен (из баллона) 23 л Комплекс хлорида алюминия 28 мл Соляная кислота, 5%-ный раствор 200 мл Хлорид кальция безводный 60 г Алкилирование бензола этиленом (пропиленом) в жидкой фазе ведут на установке, схема которой приведена на рисунке 1.1. Установка состоит из игольчатых вентилей 1, гидравлического затвора 2, склянки Тищенко 3 с концентрированной серной кислотой, трубки 4 с твердым осушителем, реометра 5, предохранительной склянки 6, контактного термометра 7, реактора 8, обратного холодильника 10, реле-регулятора 12, автотрансформатора 13 и счетчика пузырьков 14. Перед началом опыта проверяют правильность сборки, герметичность всех соединений установки, убеждаются в надежной работе реле-регулятора и возможности свободного прохождения газа по линии его сброса. Реактор изготовлен из термостойкого стекла с диаметром реакционной зоны 30 мм и высотой 200 мм. Он снабжен барботером 9, электроспиралью 11, отводной трубкой с краном 15 и краном 16 для слива реакционной массы. Алкилирование ведут при 70°С в течение 4—5 ч до поглощения реакционной массой 23 л олефина.
Рисунок 1.1 - Установка для алкилирования бензола: 1 — вентили; 2 — гидравлический затвор; 3 — склянка Тищенко; 4 — трубка с осушителем; 5 — реометр; 6 — предохранительная склянка; 7 — контактный термометр; 8 — реактор; 9 —.барботер; 10 — обратный холодильник; 11 — электроспираль; 12 — реле-регулятор; 13 — автотрансформатор; 14 — счетчик пузырьков; 15, 16 — краны.
На место контактного термометра вставляют воронку и загружают в реактор 97,5 г сухого бензола и 28 мл комплекса хлорида алюминия. Устанавливают на место контактный термометр, подают воду в обратный холодильник и азот в установку со скоростью 50 мл/мин и включают обогрев реактора. По достижении в реакторе заданной температуры прекращают подачу азота и включают подачу этилена (пропилена). Момент включения подачи олефина принимают за начало опыта. Регулируют скорость подачи олефина таким образом, чтобы в течение всего опыта поддерживалось равенство между скоростями подачи и поглощения олефина, что контролируют по прохождению остаточного газа через счетчик пузырьков (один пузырек газа через каждые 3—5 с).
В ходе опыта каждые 30 мин отбирают 2 мл реакционной массы на анализ. Перед отбором пробы отводную трубку с краном 15 продувают воздухом с помощью резиновой груши. Пробу отбирают в градуированную делительную воронку на 10—15 мл, отделяют от катализаторного комплекса, промывают равными объемами 5%-ной соляной кислоты и воды. Органический слой сливают в колбу, сушат над безводным хлоридом кальция и анализируют на хроматографе. Опыт прекращают после подачи в реактор 23 л олефина. Выключают обогрев реактора, подачу олефина и линию олефина отсоединяют от барботера для предотвращения засасывания реакционной массы в газовую линию. Реакционную массу охлаждают до комнатной температуры, отделяют от катализаторного комплекса, переливают в делительную воронку емкостью 250 мл и обрабатывают последовательно равными объемами 5%-ной соляной кислоты и воды. Органический слой переливают в чистую колбу, сушат над безводным хлоридом кальция. После сушки органический слой отделяют от хлорида кальция, определяют его массу, анализируют на хроматографе и перегоняют. При ректификации выделяют следующие фракции: до 78°С — азеотропная смесь бензола с водой (при хорошей сушке эта фракция практически отсутствует); 78—81 °С — бензол; 81 —135°С — промежуточная смесь (смесь бензола с этилбензолом); 135—137 °С — этилбензол; выше 137 °С — остаток (принимают за полиалкилбензолы). Определяют массу и показатель преломления полученного этилбензола. Зная содержание этилбензола в продуктах реакции и массу продуктов, взятых для перегонки, вычисляют потери этилбензола при ректификации. Результаты хроматографического анализа проб реакционной массы записывают в таблицу:
где С1, С2, С3 — концентрации этилбензола, ди- и триэтилбензола соответственно, % (мол.). Коэффициенты 2 и 3 соответствуют числу молей олефина, стехиометрически необходимых для получения данного соединения.
На основании полученных данных строят графики зависимости: состав реакционной массы и селективность процесса — мольное отношение поглощенного олефина к бензолу. Принимая, что достигается равновесный состав реакционной массы
С6Н6+С6Н4R2
рассчитывают по нескольким точкам константы равновесия реакции:
Обсуждают результаты опыта и формулируют свои выводы о выполненной работе. Анализ продуктов реакции
Реакционную массу анализируют на хроматографе с детектором по теплопроводности. При расшифровке хроматограмм используют метод нормировки с поправочными коэффициентами. Для определения поправочных коэффициентов в качестве стандартного вещества принимают этилбензол. Литература 1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 156-163. 2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с. Лабораторная работа №2 Тема: АРОМАТИЗАЦИЯ Н-ПАРАФИНОВ С6—С8 (ПОЛУЧЕНИЕ ТОЛУОЛА ИЗ Н-ГЕПТАНА)
Теоретическая часть
Ароматические углеводороды, широко используемые в химической промышленности, получали ранее из жидких продуктов термической переработки твердого топлива. В настоящее время главным источником ароматических углеводородов стала нефть. Впервые возможность получения ароматических соединений из нафтеновых и парафиновых углеводородов нефти была доказана работами русских ученых Н. Д. Зелинского, В. Н. Ипатьева, Б. Л. Молдавского, Б. А. Казанского и др. В качестве катализаторов процесса ароматизации были предложены металлы (Pt, Pd, Ni и др.), оксиды металлов (А12О3, Сг2О3, МоО3 и др.) и их комбинации, но самыми активными оказались платиновые катализаторы. В современных методах переработки нефти процессы каталитической ароматизации, объединенные под общим названием: «риформинг-процессов», занимают исключительно важное место. Процессы риформинга позволяют превращать низкооктановые бензины в высокооктановые, обеспечивают широкое производство таких ценных ароматических углеводородов, как бензол, толуол и ксилолы, с одновременным получением большого количества водородсодержащего газа. Промышленные процессы риформинга как на металлических, так и на оксидных катализаторах проводят под давлением водорода до 5 МПа, при температурах 450—550 °С и объемной скорости подачи газа 0,2—5 ч -1. Давление водорода препятствует коксообразованию и быстрой дезактивации катализатора. Цель работы Изучение дегидроциклизации и ароматизации «-парафина и составление материального баланса опыта. Выполнение работы Реактивы Азот (из баллона) н-Гептан, 20 г Катализатор промышленный алюмоплатиновый или алюмохромовый 70 см3
Литература 1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 86-89. 2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с.
Лабораторная работа №3 УГЛЕВОДОРОДОВ Теоретическая часть
К процессам изомеризации относятся все реакции, приводящие к изменению структуры углеводородов без изменения числа и вида атомов в их молекулах. Эти реакции занимают важное место в химической технологии для производства изопарафинов из нормальных парафинов, циклогексана и его производных из других циклопарафинов, п- и о -ксилолов из менее ценного м -кси-лола и др.
Хотя реакция изомеризации парафинов является равновесной
н-С5Н12 ↔ изо-С5Н12 ↔ нео-С5Н12
но при невысоких температурах и в отсутствие катализатора она практически не протекает. Первым катализатором, который применяли для изомеризации углеводородов, был хлорид алюминия с активирующими добавками. В настоящее время широко применяют бифункциональные контакты, которые получают нанесением платины или металлов платиновой группы на оксид алюминия, алюмосиликат и др. По современным представлениям механизм реакции изомеризации как при катализе с хлоридом алюминия, так и с бифункциональными контактами, является карбоний-ионным. Вначале карбоний-ионы возникают в реакционной массе при взаимодействии олефинов с катализатором. Олефины, в свою очередь, образуются в результате расщепления или дегидрирования исходных углеводородов. Вследствие того, что процессы изомеризации парафинов сопровождаются реакциями расщепления, алкилирования и полимеризации, в продуктах изомеризации всегда присутствуют и низкомолекулярные и высокомолекулярные вещества.
Цель работы Изучение реакции изомеризации циклогексана или метилциклопентана в присутствии хлорида алюминия (активатор — хлорид водорода), составление материального баланса опыта, определение селективности процесса и вычисление константы равновесия. Реактивы Циклогексан (или метилциклопентан), 84,2 г Хлорид алюминия 15,2 г Хлорид водорода (из генератора) Соляная кислота, 5%-ная Карбонат натрия, 5%-ный раствор Хлорид кальция (безводный)
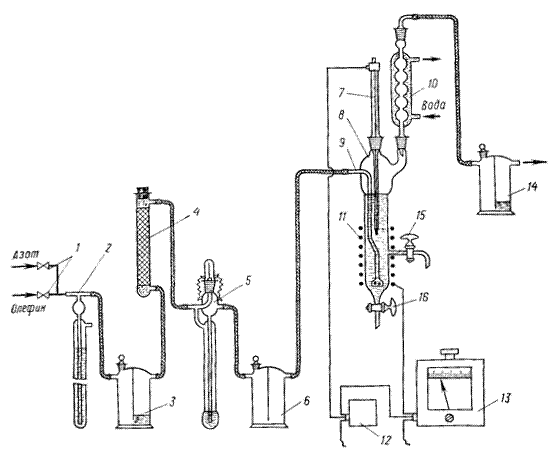
Методика проведения эксперимента Изомеризацию циклогексана в метилциклопентан проводят на установке, схема которой приведена на рисунке 3.1. Установка состоит из автотрансформаторов 1 и 11, электронного реле 2, электромотора 3, контактного термометра 4, реактора 5, электроспирали 6, крана для слива реакционной массы 7, обратного холодильника 8, мешалки 9 и крана для отбора проб 10. Перед началом опыта проверяют правильность сборки, надежность и герметичность всех соединений установки. Убеждаются в правильном присоединении электроспирали 6 и контактного термометра 4 к электронному реле 2, а электронного реле к автотрансформатору 1. На контактном термометре устанавливают температуру, которую необходимо поддерживать в реакторе 5. Проверяют работу мешалки 9, подают воду в обратный холодильник 8 и только после этого приступают к выполнению опыта.
Реактор 5 представляет собой цилиндрический сосуд из термостойкого или кварцевого стекла с реакционным объемом 150 мл. Он снабжен тремя горловинами для установки контактного термометра 4, обратного холодильника 8 и механической мешалки 9, электроспиралью 6, краном 10 для отбора проб и краном 7 для слива реакционной массы. В сухой чистый реактор через горло для контактного термометра загружают 84,2 г циклогексана, 15 г хлорида алюминия, вставляют в горло для контактного термометра барботер, включают мешалку на слабое перемешивание и насыщают реакционную массу хлоридом водорода в течение 30 мин.
Рисунок 3.1 - Установка для изомеризации циклогексана: 1, 11 - автотрансформаторы; 2 — электронное реле; 3 — электромотор; 4 — контактный термометр; 5 — реактор; 6— электроспираль; 7 — кран для слива реакционной массы; 8 — обратный холодильник; 9 — мешалка; 10 — кран для отбора проб.
Отходящие газы через обратный холодильник 8 отводят под тягу или поглощают холодной водой. После насыщения реакционной массы хлоридом водорода выключают генератор хлорида водорода, вместо барботера устанавливают контактный термометр, увеличивают число оборотов мешалки до умеренного перемешивания реакционной массы и включают обогрев реактора. С помощью автотрансформатора 1 подают на контакты нагревательного элемента реактора такое напряжение, чтобы перепад температур между спиралью нагревательного элемента и реакционной массой был небольшим. За начало опыта принимают момент закипания циклогексана. Опыт ведут при умеренном кипении реакционной массы в течение 3 ч и за это время отбирают семь проб для анализа. Первую пробу отбирают в начале опыта, а затем каждые 30 мин. Объем одной пробы не должен превышать 2 мл. Пробу переносят в делительную воронку емкостью 20 мл, обрабатывают последовательно 5 мл 5%-ного раствора соляной кислоты, 5 мл 5%-ного раствора Na2CO3 и 5 мл воды. Органический слой отделяют от воды, сушат над безводным хлоридом кальция и анализируют на хроматографе на содержание циклогексана, метилциклопентана и других углеводородов. По окончании опыта выключают обогрев реактора и охлаждают реакционную массу до комнатной температуры. Устанавливают на место контактного термометра капельную воронку и осторожно при перемешивании к реакционной массе приливают 5%-ный раствор соляной кислоты до образования двух прозрачных жидких фаз. Реакционную массу переносят в делительную воронку, органический слой отделяют от водного, промывают равными объемами 5%-ного раствора Na2CO3 и воды, сушат над безводным хлоридом кальция и определяют массу. При составлении материального баланса опыта учитывают массу проб, которые были взяты для анализа. На основании полученных данных строят графики зависимости: концентрация циклогексана — время; концентрация метилциклопентана — время (эти два графика представляют на одном рисунке); селективность — степень конверсии циклогексана. Вычисляют константу равновесия изомеризации циклогексана, обсуждают полученные зависимости и формулируют свои выводы о выполненной работе.
Получение хлорида водорода
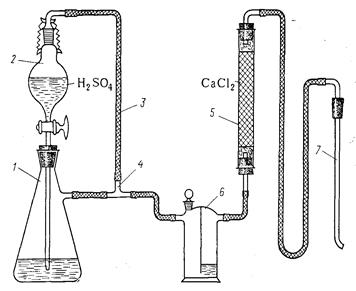
Хлорид водорода получают в приборе, изображенном на рисунке 3.2, который стоит из капельной воронки 2 емкостью 150 мл, колбы с боковым отводом 1 ёмкостью 700 мл, уравнительной линии 3, тройника 4, трубки с безводным хлоридом кальция 5, склянки Тищенко 6 и барботера 7. Внимание! Обязательно надеть резиновые перчатки и защитные очки! Для получения хлорида водорода в колбу 1 наливают 200 мл концентрированной соляной кислоты (d
Рисунок 3.2 – Прибор для получения хлорида водорода: 1— колба; 2 — капельная воронка; 3 — уравнительная линия; 4 — тройник; 5 — трубка с хлоридом кальция безводным; 6 — склянка Тищенко; 7 — барботер. Литература 1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 90-94. 2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с. 3. Паушкин Я.М., Вишнякова Т.П., белов П.С. Практикум по нефтехимическому синтезу. М.. Химия, 1965. 208 с. Лабораторная работа №4 ПИРОЛИЗ УГЛЕВОДОРОДОВ Теоретическая часть
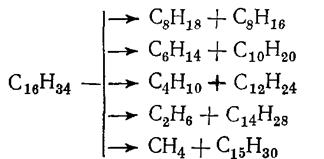
Углеводороды подвергаются наиболее глубоким химическим превращениям в процессах пиролиза, которые проводят при температурах выше 700°С. В таких условиях углеводороды подвергаются дегидрированию и расщеплению по углерод-углеродным связям с образованием газообразных, жидких и твердых продуктов. Например, первичным продуктом расщепления н-гексадекана могут быть следующие соединения, которые, в свою очередь, подвергаются дальнейшему распаду по аналогичной схеме.
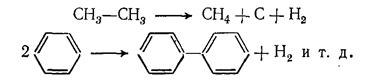
Процессы распада усиливаются с повышением температуры и времени контакта, при этом в продуктах пиролиза накапливаются вещества, более стабильные в данных условиях. При пиролизе любого углеводородного сырья в интервале 750— 850 °С целевыми веществами в газообразных продуктах реакции являются этилен, пропилен и бутилен, а в жидких — ароматические углеводороды. При дальнейшем повышении температуры пиролиза в газообразных продуктах появляется заметное количество ацетилена, усиливается выделение водорода и образование кокса, так: как выше 850 °С заметно возрастают скорости разложения углеводородов на элементы и дегидроконденсации (уплотнения) ароматических углеводородов:
В условиях пиролиза все реакции углеводородов протекают через образование свободных радикалов, которые возникают в результате расщепления связей С — С иод воздействием высокой температуры:
Свободные радикалы, в свою очередь, инициируют радикально-цепные реакции превращения углеводородов:
Пиролиз углеводородов, как правило, проводят в присутствии паров воды, что значительно смягчает условия процесса, снижает долю вторичных реакций и препятствует образованию кокса. Расход водяного пара на пиролиз колеблется от 25 до 100% от массы исходного углеводородного сырья. Цель работы Изучение влияния температуры и объемной скорости подачи исходного сырья на выход и состав газообразных и жидких продуктов пиролиза углеводородов. Реактивы н-Октан (бензин, лигроин) Азот (из баллона) Хлорид кальция (безводный)
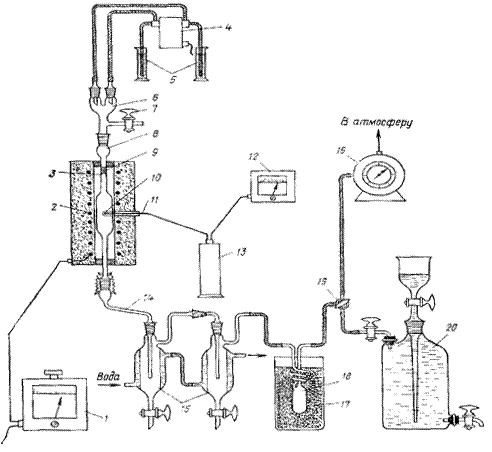
Методика проведения опыта Пиролиз н-октана проводят на установке, схема которой приведена на рисунке 4. Установка состоит из автотрансформатора 1 электропечи 2, выступов 8, дозатора 4, мерных цилиндров 5, переходников 6 и 14, крана 7, реактора 8, насадки 9, кармана для термопары 10, термопары //, милливольтметра 12, ледяных бань 13 и 17, приемников 15, газовых часов 16, ловушки 18, трехходового крана 19 и газометра 20. Перед началом опыта проверяют правильность сборки, надежность и герметичность всех соединений установки. Проверяют правильность работы электромеханического дозатора 4 для подачи в реактор 8 углеводорода и воды. Наливают в один из мерных цилиндров 5 углеводород, а в другой дистиллированную воду. Устанавливают на дозаторе заданные скорости подачи исходных веществ, отсоединяют линии подачи углеводорода и воды от переходника 6 и концы этих линий опускают в отдельные цилиндры емкостью 10 мл. Включают дозатор и по секундомеру определяют истинную скорость подачи исходных веществ; при необходимости проводят регулировку дозатора. По окончании проверки дозатор выключают, а линии подачи воды и углеводорода снова присоединяют к переходнику 6. Переключают трехходовой кран 19 на газовые часы 16 и через кран 7 продувают установку азотом со скоростью 100 мл/мин в течение 10 мин. После этого прекращают подачу азота, закрывают кран 7, включают электропечь 2 для обогрева реактора и приступают к выполнению опытов.
Рисунок 4. Установка для пиролиза углеводородов: 1 – автотрансформаторы, 2 – электропечь, 3 – выступы, 4 – дозаторы, 5 - мерные цилиндры, 6, 14 – переходники, 7 – кран, 8 – реактор, 9 – насадки, 10 - карман для термопары, 11 – термопара, 12 – милливольтметр, 13, 17 – ледяные бани, 15 – приемники, 16 – газовые часы, 18 – ловушки, 19 – трехходовой кран, 20 – газометр.
Заданием предусматривается выполнение работы по одному из следующих вариантов: 1) при постоянном массовом отношении углеводорода к воде, равном 2:1, варьируют объемную скорость подачи углеводорода (0,5, 1,0 и 2,0 ч-1) при двух уровнях температур (750 и 850 °С) — всего 6 опытов; 2) исследуют процесс методом планирования эксперимента при постоянном массовом отношении углеводорода к воде (2:1) и двух уровнях объемной скорости подачи углеводорода (1 и 2 ч -1) и температуры (750 и 850 °С) с постановкой 3 опытов в центре плана— всего 7 опытов. На один опыт расходуют 40 мл углеводородного сырья. При необходимости проверяют воспроизводимость опытов. В процессе проведения опыта наблюдают за отложением кокса на стенках реактора. При значительном его отложении перед началом нового опыта очищают стенки реактора. Для этого присоединяют к крану 7 линию азота и через установку пропускают азот со скоростью 100 мл/мин в течение 10 мин. Отсоединяют линию азота, но кран 7 не закрывают; присоединяют к приемнику 15 вместо ловушки 18 водоструйный насос и просасывают через установку воздух при 750—780°С до полного сгорания кокса. После этого отключают водоструйный насос, присоединяют к приемнику 15 ловушку 18, вновь продувают установку азотом в течение 10 мин и затем приступают к выполнению следующего опыта. Реактор представляет собой кварцевую трубку с расширенной реакционной зоной объемом около 30 мл (диаметр 20 мм). Он снабжен карманом 10 для термопары 11 и выступом 3 для удерживания кварцевой насадки 9. Высота слоя насадки 15 мм, размер частиц 4—5 мм. Заданную температуру в реакторе поддерживают с точностью ±5°С с помощью автотрансформатора / (или терморегулятора). По достижении в реакторе заданной температуры и стабилизации ее включают вначале подачу воды, а затем (через 5 мин) подачу углеводорода. Момент включения подачи углеводорода принимают за начало опыта. Продукты пиролиза через переходник 14 поступают в приемники 15, охлаждаемые водой, затем в ловушку 18, охлаждаемую ледяной баней 17, где конденсируются и собираются жидкие продукты пиролиза. Газы проходят через трехходовой кран 19, газовые часы 16 и выпускаются под тягу. Через 10 мин после начала опыта в газометр 20 отбирают 3 л газов пиролиза для анализа. По окончании опыта выключают вначале подачу углеводорода, а затем (через 5 мин) подачу воды и обогрев реактора. После каждого опыта фиксируют объем газов, прошедших через газовые часы, и объем газов, собранных в газометр. Определяют плотность и общую массу газов пиролиза, а также содержание в них водорода, этилена и пропилена. Жидкие продукты пиролиза из приемников и ловушки объединяют, отделяют от воды и сушат над безводным хлоридом кальция; находят их массу, перегоняют и анализируют. Составляют материальный баланс опыта. На основании полученных данных строит график зависимости выхода (в % масс.) газообразных и жидких продуктов пиролиза от объемной скорости подачи углеводородного сырья при двух температурах, а также выхода (в % масс.) этилена и пропилена. При исследовании процесса методом планирования эксперимента выводят регрессионные уравнения для выхода газообразных и жидких продуктов, этилена и пропилена. Обсуждают полученные зависимости и формулируют свои выводы по результатам проведенной работы. Анализ газов пиролиза
Газы пиролиза анализируют на хроматографе с детектором по теплопроводности в два приема. В одной пробе определяют только содержание водорода, в другой — содержание остальных компонентов газа. Анализ жидких продуктов пиролиза
Жидкие продукты пиролиза из всех опытов объединяют, взвешивают, добавляют к ним 1 г гидрохинона для предотвращения полимеризации непредельных и осторожно перегоняют из колбы Кляйзена. После отгонки фракции, выкипающей до 200 °С, перегонку прекращают. Определяют массу отогнанной фракции и анализируют ее на содержание непредельных и ароматических углеводородов. Остаток в колбеКляйзена относят к смолистым веществам.
Контрольные вопросы 1. Сущность процесса пиролиза углеводородов. 2. Сырье и применение продуктов пиролиза. 3. Основные и побочные реакции пиролиза.
Литература 1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 94-99. 2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с. 3. Паушкин Я.М., Вишнякова Т.П., белов П.С. Практикум по нефтехимическому синтезу. М.. Химия, 1965. 208 с.
Лабораторная работа №5 Тема: ОКИСЛЕНИЕ АММИАКА
Теоретические основы
Окисление аммиака до окиси азота является начальной стадией производства азотной кислоты. Наилучшим избирательным действием на процесс окисления аммиака до окиси азота обладает платина, которая при высоких температурах может давать выход до 99 %. При проведении процесса на платиновых катализаторах окисление аммиака начинается уже при 145°С с получением главным образом элементарного азота и закиси азота. Наибольший выход окиси азота 95-98 % достигается при температурах 850-950 ° С. При повышении температуры возрастают потери платины, что приводит к необходимости ограничить несколько верхний температурный предел процесса. Повышение температуры выше 1 000°С приводит к побочным процессам. Чрезмерное повышение температуры исходного газа сопровождается диссоциацией аммиака и окиси азота. Оптимальные температуры окисления аммиака на платине при атмосферном давлении находятся в интервале 800-840°С; для окисленных катализаторов оптимальные температуры несколько ниже и составляют 700-800 °С. В качестве окисляющего агента для процесса окисления аммиака используется в подавляющем большинстве кислород воздуха. Расход кислорода на окисление аммиака до окиси азота может быть определен согласно уравнению:
4NH3 +5O2 → 4NO+6H2О
Таким образом, стехиометрическое мольное соотношение кислорода к аммиаку O2/NH3 составляет 1,25. Однако при таком соотношении выход окиси азота незначителен. Для его повышения необходим определенный избыток кислорода, от которого будет зависеть и концентрация аммиака в воздушно-аммиачной смеси. В производственных условиях содержание аммиака в воздушно-аммиачной смеси поддерживается в интервале 9,5-11,5 %, что соответствует отношению O2/NH3 - 1,7-2,0. Для окисных катализаторов, имеющих меньшую, чем платина активность, соотношение O2/NH3 должно быть более двух и соответственно концентрация аммиака в воздушно-аммиачной смеси должна поддерживаться в пределах 7,5-9,5 %. При изменении концентрации аммиака в исходной смеси следует учитывать, что воздушно-аммиачные смеси могут быть в определенном интервале взрывоопасны. Применяемые в промышленности аммиачно-воздушные смеси (9,5-11,8 %) практически не представляют опасности. Что касается аммиачно-кислородных смесей, то они воспламеняются со взрывом при любой концентрации аммиака в интервале температур 700-800 ° С. Реакция окисления аммиака до окиси азота протекает во внешне диффузионной области как на платине, так и на окисных катализаторах. Сама реакция протекает быстро, но общая скорость процесса вследствие внешнедиффузионного торможения может снижаться более чем на 90 %. Максимальной степени превращения аммиака в окись азота соответствует определенное время контакта. Практическое осуществление процесса окисления аммиака требует таких условий, которые обеспечивают максимальное протекание основной реакции и минимальной потери аммиака в виде молекулярного азота. С этой целью окисление аммиака проводят в присутствии активного, избирательно действующего катализатора, в сравнительно узком интервале температур, при строго определенных времени соприкосновения газа с катализатором и начальном составе аммиачно-воздушной смеси. Наиболее распространенным промышленным катализатором процесса является платина или ее сплавы с родием и палладием. Для обеспечения большой поверхности катализатора платиновые катализаторы обычно используются в виде сеток из проволоки диаметром 0,045-0,09 мм, причем свободное сечение сетки составляет 50-60 %. Достаточно полный контакт газа с катализатором достигается установкой в аппарате последовательно трех сеток. Из всех металлов платиновой группы наиболее оптимальными являются добавки родия. Сплавы платины с родием характеризуются не только более высокой каталитической активностью, чем чистая платина, но имеют лучшие прочностные показатели. Особенно это важно при работе в области высоких температур, так как у них точка плавления сплава выше, чем у платины. Применяются в промышленности также палладиевые катализаторы, несколько уступающие платинородиевым по механической прочности, но имеющие высокую каталитическую активность. Различные сплавы платины и палладия с добавками родия, серебра, иридия, кобальта, вольфрама и др. обеспечивают выход окиси азота 96-99 %. Из неплатиновых катализаторов окисления аммиака наиболее активными оказались катализаторы на основе оксидов железа и кобальта; активированные добавки хрома, марганца, висмута, никеля и др.; многие из этих катализаторов показали высокую активность. Так, на окисножелезном катализаторе, промотированном окислами висмута и марганца, степень конверсии достигает 94 %. Однако, для всех окисных катализаторов наблюдается потеря активности во времени. Для систем, работающих при атмосферном давлении, применяется двухступенчатое окисление аммиака: 1-ая ступень платинородиевая сетка, 2-ая ступень - слой таблетированного окисного (чаще железо-хромового) катализатора, толщиной 50-65 мм. Общий выход окиси азота в таких аппаратах 96,5 %, затраты на платину в этом случае сокращаются в 3 раза, снижаются и потери платины. Все окисные катализаторы относятся к типу осажденных и готовятся совместным осаждением гидроокисей из растворов серно- и азотнокислых, хлористых солей. Неплатиновые катализаторы на носителях практически не применяются вследствие малой активности. Согласно теории диффузионной кинетики окисления аммиака. Скорость процесса его окисления на платине определяется наиболее медленной стадией - диффузией аммиака к поверхности катализатора. Продолжительность реакции окисления аммиака на платине составляет 10 -4-10 -5 сек. Время контактирования может быть определено из уравнения:
где V св - свободный объем катализатора, м3 /сек; Vr - объемная скорость газа в условиях конверсии, м3 /сек.
Существует вполне определенное время контакта и соответственная ему скорость газового потока, обеспечивающие максимальный выход окиси азота. Для осуществления реакции необходимо соответствующее энергетическое состояние реагирующей системы, определяемое значениями энергии активации. Процесс каталитического окисления аммиака начинается со стадии активированной адсорбции кислорода на поверхности катализатора с образованием промежуточного соединения, затем происходит активированная адсорбция аммиака, требующая меньшей энергии активации; при этом образуется переходной комплекс с последующей перегруппировкой его в комплекс. Возможно образование других промежуточных соединений гидроксиламина NH2OH, нитроксила NHO, амида NH2, атомарного азота и др. и в конечном счете образование NO, N2, H2O. Из всех термодинамически возможных реакций в присутствии катализатора в первую очередь протекает та реакция, которая требует наименьшей энергии активации. В соответствии с адсорбционно-химической теорией катализа механизм каталитического окисления на платине можно представить следующим образом. Кислород и аммиак диффундируют из потока к поверхности катализатора. Находящиеся на поверхности платины атомы со свободными валентностями обеспечивают первоочередную активированную адсорбцию кислорода за счет возникновения электронной связи. Химическое взаимодействие платины с кислородом приводит к ослаблению атомарных связей в молекулах и образованию перекисного комплекса катализатор - кислород. Последующая активированная адсорбция аммиака дает в результате комплекс катализатор-кислород-аммиак и в конечном результате происходит перераспределение электронных связей и соединение атомов азота и водорода с кислородом. Адсорбированный кислород не входит в кристаллическую решетку платины и образует с ней непрочные связи. Молекулы аммиака ориентируются к кислороду атомами водорода (вследствие высокого сродства кислорода и водорода) с последующим образованием молекул окиси азота и воды. NO и Н2О свойственна малая адсорбционная способность; вследствие этого они десорбируются с поверхности катализатора, освобождая связи для вновь сорбируемых молекул кислорода. Механизм окисления аммиака на окисных катализаторах, по-видимому, не будет иметь принципиального отличия от такого на платине; однако, оптимальные условия процесса (температура, состав газа, время контактирования) в этом случае изменяются в зависимости от стадии, лимитирующей процесс в целом. Целью данной работы является испытание активности неплатиновых катализаторов на лабораторной модельной установке в зависимости от условий: температуры, объемной скорости газа, его начального состава, времени контактирования и т.д. Методика проведение опыта
Контактный аппарат представляет собой кварцевую или фарфоровую трубку с катализатором 1, установленную в вертикальной (или горизонтальной) трубчатой печи 2 с электрообогревом. Температура в контактном аппарате заменяется термопарой — 3 и регулируется автоматическим терморегулятором 4. В кварцевую трубку помещён неплатиновый гранулированный катализатор. Трубка имеет длину около 300 мм и внутренний диаметр 20-30 мм. Газ подается в контактный аппарат сверху через стеклянную трубку 5. Газовая смесь готовится следующим образом: аммиак из склянки 6 через реометр 7 подается для смешения в стеклянный или алюминиевый смеситель 8 с воздухом, подаваемый воздуходувкой 9 через реометр 10. Из смесителя газ подается в контактный аппарат и проходит в нем сверху вниз. Для поглощения окислов азота газ после контактного аппарата пропускается через поглотительную склянку 11 с раствором перекиси водорода и отводится далее в тягу. Опыт проводится в следующем порядке: расходы аммиака и воздуха, требуемые для получения смеси заданного состава, устанавливают по заранее градуированному реометру. Общая объемная скорость газовой смеси задается в пределах 200-1000 см3/мин. Состав газа устанавливают и контролируют путем анализа. Газовая смесь с определенной заданной скоростью подается в контактный аппарат, нагретый до заданной температуры. Температура задается в пределах 60
|
|||||||||
|
|
Последнее изменение этой страницы: 2020-12-09; просмотров: 492; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 52.14.85.76 (0.187 с.) |
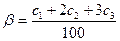
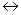 2С6Н5R
2С6Н5R

 =1,198), устанавливают на колбе воронку 2, закрывают кран и наливают в воронку 100 мл концентрированной серной кислоты (d
=1,198), устанавливают на колбе воронку 2, закрывают кран и наливают в воронку 100 мл концентрированной серной кислоты (d 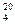 =1,836). Присоединяют к воронке уравнительную линию 3, а барботер 7 креактору изомеризации таким образом, чтобы конец барботера доходил до дна реактора. В склянку Тищенко наливают концентрированную серную кислоту до уровня на 2—3 мм выше верхнего среза отверстия в перегородке и только после этого осторожно приливают серную кислоту к соляной кислоте с такой скоростью, чтобы можно было считать пузырьки газа, проходящие через склянку Тищенко.
=1,836). Присоединяют к воронке уравнительную линию 3, а барботер 7 креактору изомеризации таким образом, чтобы конец барботера доходил до дна реактора. В склянку Тищенко наливают концентрированную серную кислоту до уровня на 2—3 мм выше верхнего среза отверстия в перегородке и только после этого осторожно приливают серную кислоту к соляной кислоте с такой скоростью, чтобы можно было считать пузырьки газа, проходящие через склянку Тищенко.