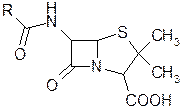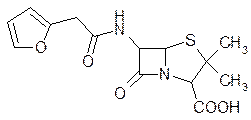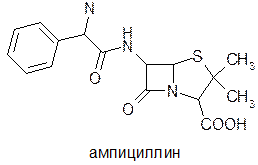Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Синтез сульфопроизводных на основе 3н-бензоксазол-2-она ⇐ ПредыдущаяСтр 8 из 8
Синтез 3Н-бензоксазол-2-она 5 осуществлялся взаимодействием 2-аминофенола 4 с мочевиной в присутствии соляной кислоты при нагревании (схема1) [5].
Спросить, что за реакция – нуклеофильное замещение. Реакция идет с отщеплением аммиака, кот. Связывается HCl. 12 часов при кипячении соотношение аф:моч: HCl=1:1.2:4 Найти спектр
Сх б-на Реакция электрофильного замещения ориентанты первого и второго рода, куда пойдет сх? Система активирована, поэтому условия сх мягкие. Р-я сх идет в два этапа Необходимо отделить от воды – экстракция. – Позволяет отделить от воды и от сульфокислоты. Очистка – флэш-хроматография. Возможна перекристаллизация. Сущность того и другого. 2-оксо-2,3-дигидро-бензоксазол-6-сульфонилхлорид (6): К 40 мл хлорсульфоновой кислоты (0.5 моль), охлажденной до 10 °С, при интенсивном перемешивании порциями добавляли 13.5 г (0.1 моль) 5 в течение 30 минут. Реакционную смесь перемешивали при 20 °С в течение 20 минут, затем нагревали до 60 °С и перемешивали в течение 2 ч. После охлаждения реакционную массу выливали на 300 г измельченного льда. Продукт отфильтровывали, промывали водой и экстрагировали в 300 мл диэтилового эфира. Органический слой промывали водой и сушили над хлористым кальцием, растворитель отгоняли. Продукт перекристаллизовывали из толуола. Выход 6 – 10.5 г (45%), т. пл 188-189 °С. Модификация б-на Метилирование: р-я нуклеофильного замещения Депрототнирование происходит легко. Реакцию проводят в ДМФА в присутствии поташа. Т- Дальше – сульфохлорирование: 3-Метил-2-оксо-2,3-дигидро-1,3-бензоксазол-6-сульфонилхлорид (10): К 20 мл хлорсульфоновой кислоты (0.3 моль), охлажденной до 30-40 °С, при интенсивном перемешивании порциями в течение 30 минут добавляли 14.9 г (0.1 моль) 9. Реакционную смесь перемешивали при 40 °С в течение 20 минут. Затем добавляли 25 г PCl5 (0.12 моль) при 50-70 °С в течение 1 ч. Реакционную массу нагревали до 80 °С и перемешивали в течение 1 ч. После охлаждения реакционную массу выливали на 500 г измельченного льда, продукт отфильтровывали, промывали водой и экстрагировали в 100 мл хлороформа. Экстракт пропускали через силикагель, отгоняли хлороформ под вакуумом, и сушили. Перекристаллизовывали из толуола. Выход соединения 10 – 7.5 г (51%), т. пл. 159-160 °С. Добавлением PCl5 можно добиться снижения расхода ХСК, смягчения условий на стадии хл-я.
Доказательство положения сульфохлорирования проведено на примере:
Рис. 3. Структура соединения 11е.
Рис. 4. Двумерный корреляционный спектр 1Н-1H NOESY соединения 11е.
В 1Н-1Н NOESY спектре соединения 11е наблюдается кросс-пик, характеризующий взаимодействие протона группы NCH3 (хим. сдвиг 3,4 м.д.) с одним ароматическим протоном (протон при 4С, 7,4 м.д.). Сигнал этого протона представляет собой дублет с константой J=8,3 Гц, характеризующей его орто-взаимодействие с еще одним протоном (протон при 5С, 7,7 м.д.). Последнее было бы невозможным в случае местонахождения сульфогруппы в положении 5-. Одновременно в пектре NOESY наблюдается кросс-пик, характеризующий взаимодействие этих двух протонов. Кроме того, на спектре присутствует кросс-пик, характеризующий взаимодействие протонов при 5С и 7С (7,5 м.д.) с протонами CH3 групп аминного фрагмента (1’С, 1,05 м.д.), причем сигнал протона при 7С представляет собой синглет.
Алкилирование б-на эфиром хлоруксусной кислоты. Так же нуклеофильное замещение, поташ, 80 гр, 2 часа. Очистка – крист. Сульфохлорирование: Этиловый эфир (6-хлорсульфонил-2-оксо-бензоксазол-3-ил)-уксусной кислоты (15): К 20 мл (0.3 моль) хлорсульфоновой кислоты, охлажденной до 10 °С, при интенсивном перемешивании порциями добавляли 23.3 г (0.1 моль) 14 в течение 30 минут. Реакционную смесь перемешивали при 30 °С в течение 30 минут. Затем добавляли 25 г (0.12 моль) PCl5 в течение 1 ч постепенно повышая температуру с 30 до 50 °С. Затем реакционную смесь нагревали до 60 °С и перемешивали в течение 2 ч. После охлаждения реакционную массу выливали на 300 г измельченного льда. Продукт отфильтровывали, промывали водой и экстрагировали в 100 мл хлороформа. Органический слой промывали водой, сушили над хлористым кальцием, затем растворитель отгоняли под вакуумом. Продукт перекристаллизовывали из толуола. Выход соединения 15 – 15.4 г (48%), т. пл. 136-137 °С. Дальше реакция ацилирования амина сульфохлоридом.
Дальше – гидролиз эфира. Возможны два типа гидролиза сложных эфиров – щелочной и кислотный. Щелочной рушит цикл. В кислоте цикл устойчив.
Общая методика получения кислот 12, 12’, 17, 17’: К раствору, содержащему 0.1 моль эфира кислоты (11а, 11б, 16, 16’ соответственно) в 200 мл диоксана добавляли при перемешивании 200 мл соляной кислоты (36%). Реакционную смесь нагревали до 100 °С и перемешивали в течение 2 ч. Затем охлаждали до комнатной температуры. Продукт отфильтровывали, промывали водой и перекристаллизовывали из изопропилового спирта. Этиловый эфир (6-хлорсульфонил-2-оксо-бензоксазол-3-ил)-уксусной кислоты (15): К 20 мл (0.3 моль) хлорсульфоновой кислоты, охлажденной до 10 °С, при интенсивном перемешивании порциями добавляли 23.3 г (0.1 моль) 14 в течение 30 минут. Реакционную смесь перемешивали при 30 °С в течение 30 минут. Затем добавляли 25 г (0.12 моль) PCl5 в течение 1 ч постепенно повышая температуру с 30 до 50 °С. Затем реакционную смесь нагревали до 60 °С и перемешивали в течение 2 ч. После охлаждения реакционную массу выливали на 300 г измельченного льда. Продукт отфильтровывали, промывали водой и экстрагировали в 100 мл хлороформа. Органический слой промывали водой, сушили над хлористым кальцием, затем растворитель отгоняли под вакуумом. Продукт перекристаллизовывали из толуола. Выход соединения 15 – 15.4 г (48%), т. пл. 136-137 °С.
Дальше – удлинения с бок-пиперазином. Так же кислотный гидролиз.
Способы синтеза виагры В настоящее время известны три коммерческих ингибитора фосфодиэстеразы (V) – силденафил, варденафил и тадалафил.
Первый коммерческий PDE (V) – Силденафил цитрат, являющийся активным ингредиентом «Виагры» был зарегистрирован в сентябре 1997 года. Поскольку это был первый орально вводимый препарат для лечения MED, то уже через 6 месяцев после регистрации в FDA он получил патент и был выпущен на рынок. Два других коммерческих PDE (V) – ингибитора, были на утверждении в FDA более длительное время. Эти два препарата (варденафил и тадалафил) были выпущены на рынок в США во второй половине 2003 года. Как видно из рис. 1, Варденафил является структурным аналогом Силденафила, в то время как Тадалафил является принципиально иной молекулой, базирующейся на тетрагидро-β-карбомине. Со времени выхода «Виагры» на рынок в 1998 году спрос на коммерческие ингибиторы PDE (V) постоянно растет (рис. 2) [49] и вероятнее всего, что эта тенденция будет продолжаться и в дальнейшем.
Рис.2 Данные по мировой продаже препаратов-ингибиторов фосфодиэстеразы (v) Впервые сиденафил был синтезирован по схеме:
Первоначальные условия (1990): (i) Step 1, T, SOCI2 ацетон, затем NH3 (водный) (78 %), (ii) Step 2a SnCI2, T, этанол (94 %) (iii) Step 2b (COCI)2, CH2CI2 перегонка (89 %), (iv) Step 2с, ТЭА, CH2CI2, 25°C, хроматографическая очистка (40%) (v) Step 3, NaOH, этанол, H2O2; затем экстракция смесью CH2CI2: метанол, затем хроматографическая очистка (72 %) (vi) Step 4, CISO3H, затем выделение в воду, затем экстракция смесью CH2CI2: метанол. (vii) Step 4, N-метилпиперазин, этанол (viii) Step 5, образование соли лимонной кислоты. Выход продукта в расчете на 1 – 9,8 %
Альтернативные пути синтеза: Такой метод предложила индийская компания Oprug [62]. (Синтез через реакцию Фриделя – Крафтса). Сульфамоилхлорид 19 был получен взаимодействием N-метилпиперазина и SO2Cl2. Далее этот сульфохлорид использован для взаимодействия с пиразолпиримидиноном 14 по реакции Фриделя – Крафтса.
Схема 12. Реакция Фриделя – Крафтса
Первым делом необходимо было синтезировать несколько килограммов вещества для предварительного исследования токсичности. Четыре изменения: 1. Использование стехиометрических количеств SOCI2 в растворителе, а не SOCI2 как растворитель на стадии (i). Это безопаснее и имеет меньшее экологическое воздействие. 2. Удаление восстановления SnCl2 на стадии (ii) Олово – тяж. Металл, 3. Использование SOCI2, а не оксалилхлорида на стадии (iii). Эта замена дала усовершенствование безопасности рабочего персонала, предотвращая эмиссию угарного газа. 4. Устранение перекиси водорода из синтеза. Водородный пероксид причиняет ожоги на контакте с кожей и – пожароопасен при транспортировке и особенно в контакте с органическим материами.
Химики используют восстановление хлоридом олова, в случае когда обычное восстановление водородом не было эффективно. Исследование показало, что неудача гидрирования была по причине следов соединений серы, от предыдущей стадии. Строгий контроль этих примесей, например, применение стехиометрического количества SOCI2 позволило избавиться от олова, и заменить эту стадию каталитическим восстановлением водородом. Каталитические гидрирования - среди самых чистых экологических химических стадий. В реакции преврашения nitro группы в амин, единственный побочный продукт - вода и есть возможности для регенерации растворителя и катализатора. Первоначально реакцию циклизации (4) в (5) проводили в водном растворе гидроокиси натрия и водородного пероксида с 72%-ом выходом. Исследования показали, что с использованием т-бутилата К в т-бутиловом спирте можно получить 5 с выходом 100% без заметного кол-ва примесей. Эта чистая реакция с высоким выходом стала первым шагом в разработке коммерческого маршрута.
Т.о.: Оптимизированные условия (1994) Optimised Med Chem Process (1994) (i) Step 1, SOCI2, толуол 50-60°C (92%), (ii) Step 2a H2, Pd / C, этилацетат (100%), (iii) Step 2b, SOCI2 ДМФА (cat), этилацетат (100 %) (iv) Step 2с, пиридин, этилацетат (84 %) (v) Step 3, третбутилат калия. Третбутиловый спирт (100%) (vi) Step 4, CISO3H, затем выделение в воду, затем экстракция CH2CI2 (vii) N-метилпиперазин, толуол, (71 %), перекристаллизация из 2-бутанона (80%) (viii) Step 5, лимонная кислота, ацетон (91%), перекристаллизация из водного ацетона (90 %).
Суммарный выход 35,9%
Главные идеи коммерческого пути синтеза представлены на схеме:
стратегические преимущества коммерческого маршрута были следующие:
· Большая выход · Чистая реакция cyclisation - теперь в конце пути. Потенциально опасные материалы теперь используются в начале синтеза. · Экологические проблемы с использованием HOSO2Cl (то есть большие объемы водных кислых сливов, требующих neutralisation и повышенный уровень гидролиза продукта на стадии выделения сульфохлорида в воду, уменьшены, за счет перевода реакции
Дальнейшие улучшения были предложены в рамках этой схемы
1. Использование 1 моль SOCI2 вместе с HOSO2Cl на стадии СХ ЭБК для полной конверсии СК в СХ 2. Низкая Тпл. ЭБК (19-20) позволяет использовать ее расплав и использовать сравнительно меньшие кол-ва ХСК. 3. Для упрощения полученный влажный СХ суспензируется в воде и так реагирует с с N-methylpiperazine. После реакции рН доводится до 7 и сульфамид отфильтровывается. Т.О. на этой стадии не используются органический р-ли. 4. Использование толуола в кач-ве р-ля на 1-й стадии позволило снизить к-во SOCI2 до 1,2-1,6 моль/моль. Толуол рециклизуется. 5. Восстановление в ЭА водородом. 6. Активация к-ты 6 с пом. КДИ. Преимущества КДИ · чистая химия и высокое качество продукта. · Позволяет объеденить все три реакции гидрирование, активация и · Единый растворитель, который используется (ацетат этила), · Не используются хлорсодержащие реагенты. · Высокий выход, (90 %) на три стадии
Полученый продукт циклизуют тБК в тБС. Процесс ведут при высоких конц. Для уменьшения к-ва отходов. По оконч. Р-ии добавляют воду, рН доводят до изоэлектрической точки и осадок сиденафила отфильтровывают. Продуктом явл. Лимоннокислая соль. Был разработан метод получения СЦ сочетающий высокий выход (98-100%) и высокое качество. Т.О. Условия коммерческого метода синтеза (1997): (i) Step 1, SOCI2, DMF (cat.), Т толуол; NH3 (водный) (92%) (ii) Step 3a, H2 Pd/C этилацетат (100%) (iii) Step 2, CISO3H, SOCI2 25°C; (iv) Step 2, N-метилпиперазин, вода, 25°C затем нейтрализация (v) Step 3b, 6 + CDI, этилацетат, затем + 3 (90 %) (vi) Step 4, третбутилат калия. Третбутиловый спирт (92 %) (vii) Step 5, лимонная кислота, 2-бутанон (99 %).
1997 г - Суммарный выход 75%
Как только стадии были разработаны, начались исследования по рециклизации растворителей, используемых в синтезе. Рециклизация толуола, этилацетата и 2-бутанона была введена в 1998, году, когда Viagra ™ был запущен на рынок. Как только стадии были разработаны, начались исследования по рециклизации растворителей, используемых в синтезе. Рециклизация толуола, этилацетата и 2-бутанона была введена в 1998, году, когда Viagra ™ был запущен на рынок. Использование растворителей на разных стадиях разработки процесса дается в рис. 1.
Рис. 1 количество органической траты(отходов), произведенной sildenafil солью лимонной кислоты обрабатывает в различных пунктах(точках) времени.
Из специфического примечания - полное устранение хлорированных растворителей и устранения очень летучих растворителей типа диэтилового эфира, хлорида метилена, метанола и ацетона. Устранение этих очень летучих растворителей уменьшает атмосферное загрязнение, как будет отмечено позже. Дальнейшее усовершенствование синтеза (описанного в Схеме 3) было заменой т-бутанола. Этот растворитель полностью смешивается с водой в любых соотношениях и трудно восстанавливается для повторного использования. Есть перспективы для улучшенной экологических характеристик процесса за счет перехода от т-бутанола к другому растворителю, который может быть восстановлен. Этот новый процесс был исследуется и оптимизируется. (данные на 2003 г.) и после введения его в эксплуатацию расход растворителей составит 4л/кг.
Коммерческий синтез Силденафила представлен на схеме 1 [50]. Ключевым соединением для этой схемы является нитропиразол 4, синтез которого описан в разделе 2.4. Другое ключевое соединение – сульфамид 5. Его синтез осуществляется путем последовательного сульфохлорирования исходной кислоты и амидирования сульфохлорида. Взаимодействие, приводящее к соединению 6 осуществляется когда вещества находятся в виде раствора в этилацетате в одной реакционной массе, и при этом нитропиразол подвергается каталитическому восстановлению в присутствии КДИ.
Схема 1. Коммерческий синтез Силденафил цитрата
Альтернативные пути синтеза: Во-первых конденсация альдегида 7 с аминопиразолом 8 в кипящем толуоле приводит к получению дигидросилденафила 9. Выход по данной реакции составляет 52 – 95 %. Выделение целевого продукта осуществляется путем отгонки толуола азеотропной дистилляцией и в зависимости глубины дистилляции изменяется выход. Дигидросилденафил 9 далее окисляют до Силденафила: а) с использованием катализатора Pd/С в присутствии малых количеств трифторуксусной кислоты при высокой температуре; б) либо гидросульфата натрия. Химия этой схемы описана в работах [52, 53] в случае, когда, окисляющим агентом является гидросульфат натрия, общий выход 1 в расчете на аминопиразол составляет 85 %. Условия получения 9: кипящий толуол (52 %) или кипящий толуол с последующей азеотропной отгонкой (92 %).
Схема 2. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 1)
Условие получения 1 из 9: Pd/С, трифторуксусная кислота, толуол (200 °С, 84 %) или NaHSO3, кипящий диметилацетальдегид – 90 %.
Построение пиримидинового фрагмента может осуществляться на основе нитрила 12, либо через амидин 10, либо через иминоэфир 11. Эти подходы и, соответственно, экспериментальные процедуры, были описаны в работе сотрудников фирмы Pfizer [54, 55]. Амидин 10 был получен из нитрила 12 взаимодействием его с хлорметилалюминием (полученным на основе хлорида аммония и триметилалюминия).
Схема 3. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 2)
Условия получения амидина 10: гексан, Me3Al, NH4Cl, толуол, исходное – 12, сначала 12,5 °С, затем 80 °С (58 % выход). Иминоэфир 11 получен из нитрила 12 по реакции Пиннера.
Схема 4. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 3)
Условие этой реакции: этанол, пропускание газообразного HCl, 0 °С (выход 48 %).
Третий синтез – это синтез через интермедиат 13, содержащий кислотную функциональную группу в пиридиноновом цикле
Схема 5. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 4)
Циклизация интермидиата-кислоты протекает с использованием тионилхлорида и приводит к лактону 13а, который далее взаимодействует с аммиаком, приводит к пиразолпиримидинону 1 (Силденафилу). Данные процедуры описаны в работе [56]. Условия получения 1 из 13. Синтез 13а из 13: кипячение в SO2Cl2 с последующей отгонкой SOCl2. Синтез 1 из 13а в этаноле обработка аммиаком. Следующий синтез на основе нитропиразола 4.Этот подход описан в работе [57]. Соединение 15, содержащее атом F в качестве радикала R было получено при взаимодействии 2 – фторбензоилхлорида с пиразолом 4 (выход 65 %) с последующим сульфохлорированием и сульфамидированием (75 %). Далее нитрогруппа была восстановлена с помощью SnCl2 и полученный амин принял участие в реакции циклизации. На заключительной стадии (получение 1 из 15) атом F был замещен на группу OEt.
Схема 6. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 5)
Условия получения 1 из 15: восстановитель - SnCl2, EtOH, 65 °С.
Другой вариант, также как и основная, коммерчески реализуемая схема, приводит к интермедиату 6, циклизация которого дает целевой Силденафил. Однако получение интермедиата 6 основано не на 2-этоксибензойной кислоте (в качестве исходного), а на его амидном производном 16, которое сначала подвергается сульфохлорированию и амидизации сульфохлорида, а затем уже и циклизации до Силденафила.
Схема 7. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 6)
Схема 7 описана в работе [58]. Условия циклизации 6 в 1 описаны выше – KOBu, кипячение в бутиловом спирте в течение 6 – 8 часов с выходом 95 %.
Последний вариант синтеза Силденафила, заключительной стадией которого, является реакция циклизации представлен на схеме 8.
Схема 8. Альтернативные методы получения Силденафила с финальным формированием пиримидинонового цикла (Вариант 7)
Этот вариант также является коммерчески оправданным, так как в качестве исходного используется 2-хлор(фтор)бензойная кислота, которая является более дешевой, чем этоксибензойная кислота. На последней стадии данного процесса, при использовании этанола в качестве растворителя и сильного основания для активирования циклизации, происходит одновременное протекание циклизации и замещение уходящей группы. В случае, когда Х=F, выход 1 в расчете на исходный реагент составляет 100 % [59].
Антибиотики Антибиотики—вещества природного происхождения, обладающие выраженной биологической активностью. Они получаются из растений, микробов, животных тканей, а также синтетическим путем.
Среди антибиотиков значительное место занимают пенициллины. Пенициллины оказывают выраженный эффект в отношении растущих микроорганизмов (бактерицидное действие), они поражают бактерии в фазе роста, ослабляя их клеточные стенки, а, учитывая, что для бактерий характерно необычайно высокое внутреннее давление, это приводит к разрыву клеточной стенки и уничтожению бактерии. Следует отметить, что бактерии резко отличаются от всех живых организмов, в частности, необычным составом клеточных стенок, основным компонентом которых является ацетилмураминовая кислота
Создание полимера протекает ферментативно. пенициллины выступают в качестве ацилирующих агентов по отношению к транспептидазной части фермента, причем ацилирование происходит за счет легко раскрывающегося Р-лактамного цикла. Открытие Флемингом плесени Penicillinum позволило с 1941 г. наладить ее получение в количествах, достаточных для испытания на людях. Дальнейшие усилия исследователей разных стран привели к созданию высокоэффективных штаммов пенициллинов, разработке рациональных условий глубинной ферментации, выделения и очистки пенициллинов, способов и условий их применения
Было установлено, что ядро пенициллина состоит из р-лактамтиазолидиновой бициклической системы, образовавшейся конденсацией аминокислот dl-валина и 1-цистеина.
Отдельные пенициллины отличаются друг от друга различными остатками R, от строения которых в основном зависит их различное биологическое действие. С расшифровкой строения природных пенициллинов начались работы по синтезу и модификации их структур Рядом исследователей было показано, что добавление фенилуксусной кислоты в ферментационную среду приводит к повышению выхода бензилпенициллина. Это открытие привело к разработке метода биосинтеза, заключающегося в добавлении в питательную среду различных органических кислот—предшественников, используемых плесневыми грибками для построения бокового радикала.
Так был получен феноксиметилпенициллин (пенициллин V)
который оказался терапевтически активным, кислотоустойчивым и стал первым препаратом из группы пенициллинов, пригодных для перорального применения
Ферментативный метод получения бензилпенициллина вскоре был усовершенствован—повышен выход продукта, обеспечена высокая чистота, упрощено производство и т. д. Все это позволило бензилпенициллину занять ведущее положение среди антибиотиков. Однако наряду с большими достоинствами, бензилпенициллин имеет ряд недостатков: узкий спектр действия (он активен только в отношении грам-положительных микроорганизмов), малая устойчивость к действию кислот и пенициллиназы, аллергические явления и т. д. Поворотным пунктом в создавшемся положении, позволяющим обычными методами, применяемыми в органической химии, получать новые полусинтетические пенициллины с любым ацильным остатком, оказалась возможность получения исходной 6-аминопенициллановой кислоты (6—АПК). В 1959 г. выделили в чистом виде и описали.
Ферментативный метод гидролиза бензилпенициллина оказался наиболее целесообразным и лег в основу промышленного метода получения 6-АПК. Примерно к этому времени выяснилось, что применение в практической медицине только бензилпенициллина или феноксиметилпенициллина невозможно, ибо устойчивость стафилококковых штаммов к ним стала угрожающей. Эта устойчивость не была связана только с адаптацией к ним микроорганизмов, а представляла собой селекцию форм, которые с самого начала не поддавались их воздействию в силу способности вырабатывать фермент пенициллиназу, расщепляющий b-лактамное кольцо пенициллинов [9]. Уязвимым местом в молекуле пенициллина является b-лактамное кольцо, которое легко разрывается, что приводит к исчезновению антибактериального действия. За сравнительно короткий промежуток времени была проведена большая работа, благодаря которой пенициллины приобрели новый облик и смогут дальше служить человечеству при умелой и целенаправленной организации поиска новых препаратов и усовершенствования методов их применения. Полусинтетические пенициллины получаются путем ацилирования 6-АПК обычными методами. Исходя из соответствующего ацильного остатка, можно изменить некоторые свойства получаемых пенициллинов, повысить устойчивость к кислой среде, устойчивость к пенициллиназе, расширить антибактериальный спектр действия, продлить время выделения из организма и т. д. Первыми соединениями, полученными синтетически из 6-АПК, были алкилпроизводные феноксиметилпенициллина. Изучение антибактериальных свойств этой группы соединении показало, что их активность в значительной мере зависит от величины и строения вводимых алкильных радикалов и их активность в отношении Stphylococcus aureus снижается по мере удлинения углеродной цепочки в боковом радикале [11]. В то же время замещение водорода у a-углеродного атома придает им кислотоустойчивость. Из этой группы соединений в качестве лечебных средств были отобраны два препарата—фенетициллин и пропициллин.
Вся группа a-алкилфеноксиметилненициллинов, в особенности фенетициллин, оказалась более стабильной к действию пенициллиназы, чем феноксиметилпенициллин. Сравнение антибактериальных свойств диастереоизомеров фенетициллина показало, что L-изомер более активен, чем D. Замещение водорода у a-углеродного атома феноксиметнлпенициллина ароматическими радикалами [12] привело к открытию фенбиишллина, который по своим антибактериальным свойствам похож на фенетициллин.
Он обладает сравнительно узким спектром действия и его единственное преимущество заключается в том, что после приема препарата, по сравнению с феноксиметилпеницил-лином и фенетициллином, в сыворотке крови концентрация препарата примерно в два раза больше. Близким по своим свойствам к фенбициллину оказался 3,4-дихлорметоксибензил-пенициллин, выпущенный в обращение под названием риксапен. Дойл, Нейлор и другие провели ацилирование 6-АПК различными карбоновыми кислотами, имеющими заместители у «-углеродного атома [14]. При этом было установлено, что замещение всех трех атомов водорода в метилпенициллине различными радикалами приводит к выраженной устойчивости к пенициллиназе, в особенности, в случае большого объема этих радикалов. Такое повышение устойчивости к действию пенициллиназы рядом исследователей объясняется образованием пространственных затруднений у амидокарбоновой группы, что препятствует присоединению пенициллинов к лабильным группам фермента [14]. Сравнение скорости гидролиза дифенилметилпенициллина трифенилметилпепициллином показало, что второе соединение практически не подвергается гидролизу.
Дальнейшие работы показали, что затруднения у амидокарбоновой связи могут создавать также радикалы, имеющиеся в определенных положениях ароматического кольца. Так, 2,6-диалкоксифенилпенициллины также оказались устойчивыми к действию пенициллиназы. Отличным примером является самое простое соединение этого ряда—2,6-ди-метоксифенилпенициллин (метициллин)
Метициллин по своей активности уступает бензилпени-диллину, однако он более устойчив к действию пенициллиназы и менее токсичен. Метициллин, как и бензилпеницнллин, из-за низкой устойчивости в кислой среде не пригоден для лерорального применения. Аналоги метициллпна, имеющие алкоксигруппы с большим числом углеродных атомов в радикале, менее активны, чем метициллин, однако и они сохраняют устойчивость к действию пенициллиназы [15]. Перемещение алкоксигруппы метициллина в другие положения бензольного кольца, как, например, 2,4-, 2,5- и т. д., приводит к исчезновению устойчивости к действию пенициллиназы. Введение электроотрицательных радикалов в ароматическое кольцо стабилизирует препараты в отношении к кислотам, однако активность их снижается [16]. Среди пенициллинов, являющихся производными замещенных ароматических дикарбоновых кислот, следует отметить производные фталевых кислот, которые проявляют умеренную активность, но значительную стойкость к действию пенициллиназы [17].
Были получены также аналоги пенициллина из ряда нафталинкарбоновой кислоты и ее производных. Наиболее интересный препарат этого ряда 2-этоксинафтилпеницил-лин—нашел применение в медицинской практике под назва нием нафциллин
Нафциллин в отношении некоторых штаммов кокковых бактерий в 5—6 раз активнее метициллина [19]. Из других производных следует отметить 1,3-диметок-си-2-нафтилпенпциллин, сходный по своей структуре с метициллином. Этот препарат, как и метициллнн, пространственно блокирован, благодаря чему устойчив к действию пенициллиназы.
В работах по синтезу полусинтетических пенициллинов были использованы также кислоты, содержащие гетероциклические остатки, как, например, фуран. Эти препараты оказались хорошими противомикробными веществами и не уступали по своей активности и устойчивости к действию пенициллиназы нафциллину
Из пиридинкарбоновых кислот, примененных для ацили-рования 6-АПК, наиболее активным оказался 2-пиридилпенициллин, который оказался более устойчивым к действию пенициллиназы, чем бензилпенициллин и более эффективен в отношении умеренно действующих штаммов Staph. aurens [18].
Введение алкильных радикалов в пиридиновое кольцо не привело к повышению их активности. Из других азотсодержащих гетероциклов следует отметить производные хинолина. Так; 2-хинолилпенициллин обладает повышенной активностью в отношении чувствительных и умеренно устойчивых штаммов.
По аналогии с метициллином и' нафциллином был получен 6,8-диметокси-7-хинолилпенициллин. Интересно отметить, что эти три препарата, будучи пространственно блокированы, несмотря на содержание различных систем — бензола, нафталина, хинолина — одинаково устойчивы к действию пенициллиназы. Этот факт лишний раз подтверждает значение конформации пенициллинов в вопросе их инактивации пенициллиназой.
Очень высокой антибактериальной активностью обладают пиридил- и хинолилпенициллины, содержащие карбоксильную группу в различных положениях гетероциклического кольца. Из пенициллинов, содержащих карбоксильную группу в гетероциклическом кольце, наиболее интересным оказался З-карбокси-2-хиноксалилпенициллин, динатриевая соль которого нашла применение в медицинской практике под названием хинациллин [22].
Хинациллин обладает наиболее выраженным действием в отношении Staph. aureus, причем его антимикробные свойства усиливаются с уменьшением рН среды. Он практически нетоксичен и эффективен при парэнтеральном применении. Однако хинациллин по антимикробной активности и устойчивости к действию пенициллиназы уступает ряду применяемых в настоящее время полусинтетических пенициллинов. Наиболее успешной модификацией боковой цепи пенициллинов, содержащих гетероциклические системы, оказалось введение изоксазольного кольца. Эти пенициллины обладают высокой антимикробной активностью в отношении грамположительных микроорганизмов, устойчивостью к действию пенициллиназы и более сильной связываемостью с белками сыворотки крови, чем в случае метициллина. Первый препарат этого ряда, нашедший практическое применение, был 5-метил-3-фенил-4-изоксазолилпенициллин (оксациллин), обладающий высокой антимикробной активностью, схожей с активностью нафциллина
Экспериментальные и клинические испытания хлорсо-держащих аналогов оксациллина показали, что введение атомов хлора в орто- и пара-положения фенильного радикала приводит к повышению всасываемости и антимикробной активности [24]. Из этих соединений в ряде стран нашли практическое применение 5-метил-3-(о-хлорфенил)-4-изоксазолилпенициллин (орбенин) и 5-метил-3-(о,о-дихлорфенил-4-изоксазолилпенициллин (диклоцин).
В терапевтическую практику эти пенициллины вошли благодаря ряду положительных свойств: устойчивости к кислотам, возможности применения парэнтерально и перораль-но, высокой активности в отношении устойчивых к бензил-пенициллину штаммам стафилококков, чем в случае мети-циллина и даже в некоторой степени и оксациллина [25]. Следует отметить, что эти оба препарата лишь несколько активнее оксациллина, однако их концентрация в сыворотке крови в два раза выше. Дальнейшее изучение состава культуральной жидкости привело к обнаружению пенициллина N и цефалоспорина С [26]. Изучение антимикробных свойств этих соединений показало, что несмотря на низкую по сравнению с бензилпе-нициллином антимикробную активность, они выгодно выделяются своим спектром действия. В то время как бензилпе-нициллин в основном действует на грамположительные микроорганизмы и практически не активен в отношении гра-мотрицательных, пенициллин N и цефалоспорин С действуют на грамотрицательные микроорганизмы. Расшифровка структур этих соединений показала, что в их составе имеются остатки аминокислот. С установлением этого факта развернулись работы по применению аминокислот самого различного строения для синтеза пенициллинов. Из синтезированных соединений заслуживает вниманияа-аминобензилпенициллин—амлициллин [27].
Введение электроноакцепторных заместителей в г-положение ацильного остатка пенициллина приводит к возникновению кислотостойкости и действия на грамотрицательные микроорганизмы, а введение пространственно блокирующих групп — к пенициллинам, устойчивым к действию пени-циллиназы.
|
|||||||||
|
|
Последнее изменение этой страницы: 2016-08-01; просмотров: 479; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 13.58.244.216 (0.203 с.) |





 С
С