
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Влияние на фармакокинетику возраста и характера заболевания
Фармакокинетика лекарственного вещества может меняться в зависимости от возраста и заболевания больного [31-44]. Существует несколько факторов, которые могут влиять на этапы фармакокинетики у больных пожилого возраста, делая их более чувствительными к действию психотропных веществ: • Уменьшение внутриклеточной жидкости. • Снижение процесса связывания с белками. • Уменьшение массы тканей организма. • Увеличение относительного веса жировой ткани. Сочетанное появление этих изменений повышает чувствительность к действию психотропных препаратов в пожилом возрасте. Увеличение жировой ткани повышает способность к накоплению лекарственных веществ. В результате лекарство присутствует в организме более продолжительное время. Происходит также увеличение свободной фракции лекарственного вещества при любом уровне его концентрации в крови в отношении каждого введенного миллиграмма в связи со •снижением процесса связывания с белками. Дополнительно можно отметить, что в пожилом возрасте снижается интенсивность обмена лекарственных веществ (что вызвано уменьшением печеночного кровотока, уменьшением массы печени, сокращением количества и активности CYP ферментов). Снижение функции почечного выведения, связанное с возрастом, означает большее накопление активных метаболитов и их возможное отрицательное действие. Тем не менее существует два вида возрастных изменений, которые ведут к снижению накопления лекарственных веществ в организме и их действия: уменьшение интенсивности всасывания из просвета кишечника и повышение значения рН в желудке. В целом можно сказать, что в пожилом возрасте период, необходимый для выведения препарата из организма, значительно увеличивается для тех лекарственных веществ, у которых процессу выведения предшествует значительная биотрансформация. Таким образом, у больных старшего возраста процесс выведения лекарств происходит медленнее, чем у более молодых больных. При одних и тех же дозировках пожилые больные более восприимчивы к различным побочным эффектам, связанным с уровнем дозировки/концентрации препарата [45-59]. Подобный феномен может также появляться при заболеваниях, влияющих на физиологические механизмы различных фаз фармакокинетики.
Четыре основные фазы фармакокинетики ВСАСЫВАНИЕ Клинический пример: Больной, 37 лет, с шизоаффективным расстройством получал назначения per os: тиоридазин (100 мг четыре раза в день), фенитоин (100 мг четыре раза в день), амитриптилин (50 мг четыре раза в день). Клиническое состояние больного при таком режиме назначений было стабильным на протяжении четырех недель, но затем больной стал жаловаться на седацию в дневное время. Поэтому лечащий врач изменил рекомендации, назначив все три препарата в один прием перед сном. В первую же ночь после изменения режима приема у больного возникла острая остановка сердца. Этот клинический случай демонстрирует возможность кумулятивного фармакодинамического действия — замедление внутрисердеч-ной проводимости из-за выраженного повышения уровней концентрации в результате приема лекарственных препаратов одновременно. Основным способом приема психотропных препаратов является пероральный. Процесс всасывания начинается в просвете тонкого кишечника. Лекарственное вещество затем поступает в портальный кровоток и доставляется в печень. Метаболизм лекарственных веществ СYP ферментами может происходить до поступления в систему общего кровотока в стенке кишечника или в печени (так называемый первичный метаболизм). Большинство психотропных препаратов отличается выраженными липо-фильными свойствами, поэтому они легко преодолевают гематоэнцефалический барьер и поступают в центральную нервную систему [16,17]. Дополнительно в связи с этой высокой липофильностью для них характерны следующие общие свойства: • Быстрое всасывание. • Полное всасывание. • Быстрое и обширное распределение в различных тканях. • Интенсивный первичный метаболизм. • Большой объем распределения. Понятие биодоступности относится к той части лекарственного вещества, которая абсорбировалась из места введения. Эталонным для выяснения биодоступности является внутривенный способ введения, поскольку при этом происходит 100%-е всасывание. На рис. 3.2 показаны три варианта динамики концентрации лекарственного вещества. Площадь под кривой на графике представляет общее количество препарата в кровотоке, доступное для поступления в место действия. Сплошная кривая демонстрирует динамику концентрации препарата при пероральном приеме; пунктирная кривая соответствует внутримышечному введению; точечная кривая показывает динамику концентрации при внутривенном введении. Полное всасывание одной и той же дозировки при любом из этих способов введения выражается на графике одинаковой площадью под кривыми концентрации (100%-я биодоступность).
Рис. 3.2. Динамика концентрации одного и того же лекарственного препарата в плазме крови больного, назначенного однократно при различных способах введения: перорально (сплошная ли ния); внутримышечно (пунктирная); внутривенно (точечная). Любое уменьшение величины этого участка, располагающегося под кривыми внутримышечного или перорального введения, по отношению к участку под кривой внутривенного введения будет означать снижение биодоступности, связанное с путем введения препарата [5]. Основными факторами, влияющими на биодоступность, ЯВЛЯЮТСЯ: • Физико-химические свойства вещества. • Форма выпуска лекарственного препарата. • Болезненное состояние, которое может влиять на желудочно-кишечные функции или на первичный метаболизм. • Преципитация лекарственного вещества в месте инъекционного введения. Существуют и другие клинически важные параметры, связанные с динамикой концентрации однократной дозировки препарата, но не имеющие отношения к абсорбции. Они включают: • Пик концентрации (Кmax). • Время, необходимое для достижения пика концентрации (Тmax). В общем, kmax обратно коррелирует с Т max (т.е. чем меньше необходимо времени для всасывания лекарственного вещества, тем выше будет максимальная концентрация препарата). Чем выше Кmax и чем короче Тmax, тем быстрее проявляется действие препарата, следующее за его введением. Кmax и Тmax, в целом отражают физико-химические свойства химического соединения, что может определять соответствие разрабатываемого лекарства определенным показаниям. Замедленное всасывание из просвета кишечника и медленное проникновение в ткань головного мозга из общего кровотока более характерны для веществ с высокой полярностью. Обычно существует корреляция между этими двумя этапами (поступление вещества в общий кровоток и поступление в головной мозг). Оксазепам, наиболее полярный бензодиазепин, очень медленно абсорбируется в общий кровоток, а также и в ткань мозга [8, 9]. Подобные фармакокинетические свойства не всегда согласуются с его применением как седативно-сно-творного средства. В противоположность этому лоразепам быстро проникает как в кровоток, так и в мозговую ткань и поэтому достаточно быстро может вызвать сон [17]. Скорость проникновения в мозговую ткань обычно соответствует скорости проникновения в общий кровоток, но иногда они могут и не совпадать. Примером может служить темазепам с его необычной формой выпуска в виде желатиновой капсулы, которая не расщепляется под действием желудочного сока [8]. При такой форме выпуска всасывание происходит медленно, снижая, таким образом, выраженность седативного действия этого препарата, несмотря на то что он достаточно быстро поступает из общего кровотока в мозг. В другой форме выпуска его всасывание может происходить намного быстрее, повышая тем самым его эффективность как седативно-снотворного средства.
Быстрое всасывание не всегда желательно, поскольку осложнения могут быть связаны со значением Ктах. Клинический пример, приведенный в начале нашего обсуждения абсорбции, показывает, как незнание этого факта привело к непоправимым последствиям. Кардио-токсичность, связанная с электрофизиологической стабилизацией мембран, которая настолько же зависит от высокой концентрации препарата в сыворотке крови, насколько и от уровня устойчивой концентрации в ткани. Таким образом, можно значительно повысить безопасность препарата, изменяя форму выпуска, для задержки Ттах и снижения Кmax. Той же цели можно достичь, назначая препарат в дробных дозировках при частом приеме. В этом случае средний уровень концентрации препарата в сыворотке крови между приемами и количество всасываемого вещества остаются неизменными, но значение пиковой концентрации будет ниже, а минимальной — выше. Различия в биодоступности, которые могут быть связаны со скоростью всасывания, весьма значительны между разными формами выпуска одного и того же препарата. Комиссия по пищевым и лекарственным препаратам считает генерическую (непатентованную) форму препарата сопоставимой с фирменным продуктом, если она отличается не больше чем на ±20% по биодоступности (например, Тmax и Ктах) [60]. Следовательно, теоретически между двумя генерическими формами одного препарата может быть разница до 40%. Возможно, это является объяснением того, почему у больного появляются побочные эффекты или возникает обострение болезни при назначении другой формы препарата, который в прошлом давал положительные результаты. Способ введения лекарства Способ введения лекарственного вещества может влиять на скорость его всасывания, а также на соотношение между исходным веществом и его различными метаболитами. Например, кривая концентрации при внутримышечном введении обычно смещена влево в связи с более быстрым всасыванием. Следовательно, Тmax сдвинуто влево (т.е. сокращено), а Кmax становится выше, круче изгибая кривую, несмотря на то что общая величина участка, находящегося под кривой, остается неизменной.
Тем не менее подобная модель не обязательна для всех лекарств. Например, диазепам и хлордиазепоксид при значении рН =7,4 становятся неустойчивыми и при внутримышечном введении кристаллизуются в тканях [8, 9, 17]. Таким образом, их биодоступность ниже при внутримышечном введении, чем при приеме per os. Процессу их абсорбции свойственна неустойчивость и изменчивость в зависимости от места их инъекционного введения (вблизи кровеносных сосудов, в жировой или мышечной ткани). В подобном случае их всасывание бывает неполным и медленным. Первичный метаболизм При пероральном приеме всасывание лекарства обычно происходит в тонком кишечнике. Затем лекарственное вещество поступает в портальный кровоток и проходит через печень. CYP ферменты в стенке кишечника и в гепатоцитах могут метаболизировать определенную часть лекарственного вещества до того, как оно поступит в общий кровоток (эффект первичного метаболизма). Величина этого эффекта может зависеть от различных заболеваний (циррозы, хронические гепатиты, портокавальный анастомоз, сердечная недостаточность) и фармакологически активных веществ (алкоголь, кетаконазол, флуоксетин), которые влияют на уровень концентрации препарата и на соотношение изначального вещества с его активными метаболитами [57,58]. Метаболиты, образованные в результате этого первичного эффекта, выделяются с желчью в просвет тонкого кишечника. Жирорастворимые вещества повторно всасываются в печеночный кровоток и в конце концов поступают в общую систему кровообращения. Профиль фармакологической активности этих метаболитов может почти соответствовать или же существенно отличаться от профиля изначального лекарственного вещества. Так, например, процесс биотрансформации хлорпромазина в печени теоретически ведет к появлению 168 метаболитов, 70 из которых обнаруживаются в сыворотке крови или в тканях. Некоторые из них обладают свойством блокировать дофаминовые рецепторы, хотя и намного слабее. Это затрудняет возможность различать терапевтическое действие собственно изначального лекарственного вещества и его активных метаболитов. При внутривенном или внутримышечном введении, в отличие от перорального приема, лекарственное вещество поступает непосредственно в общую систему кровообращения, не подвергаясь при этом первичному метаболизму. Поэтому для многих лекарств (например, флуфеназин) внутримышечные назначения являются более сильнодействующими, и это всегда необходимо учитывать в расчете дозировки препарата при парентеральном способе введения. При некоторых заболеваниях, таких как цирроз печени, возникает явление портокавльного шунтирования, при котором лекарство также поступает непосредственно в общий кровоток, не подвергаясь первичному метаболизму, что усиливает его психотропное действие. Сочетанное назначение нескольких лекарственных средств также может влиять на процесс первичного метаболизма [61-63]. Например, алкогольная интоксикация ведет к существенному уменьшению первичного метаболизма ТЦА и соответственному двукратному повышению концентрации этих антидепрессантов в крови [64]. Этим объясняется повышенная токсичность высоких дозировок трициклических антидепрессантов на фоне приема алкоголя.
РАСПРЕДЕЛЕНИЕ Клинический пример: У 44-летнего мужчины, больного алкоголизмом, доставленного в приемный покой, наблюдались явления дезориентировки и агрессивности после двухдневного состояния абстиненции. Он жаловался на зрительные и тактильные галлюцинации. У больного определялись тахикардия, повышение артериального давления и высокая температура. Был назначен лоразепам внутривенно капель-но. Первые признаки незначительной седации появились спустя 15 мин. После этого больной был переведен в стационарное отделение. При переводе в отделение в течение 30 мин был прерван прием лоразепама, и в результате возобновилась прежняя симптоматика. Он был возбужден и проявил агрессию по отношению к одной из медсестер. Больного вынуждены были фиксировать. В приведенном случае не было учтено явление перераспределения лекарственного вещества. Это привело к осложнениям, которых можно было избежать. Лекарственное вещество, находясь в общем кровотоке, распределяется по органам пропорционально содержанию в них жиров и белков [65]. Скорость накопления вещества зависит от развитости микроциркуляторного русла в данном органе или ткани. Хотя величина накопления жирорастворимых соединений в жировой ткани и веществе головного мозга одинакова, скоростьнакопления намного выше в головном мозге, чем в жировой ткани.
Это имеет особенно важное значение при внутривенном введении первичной дозировки психотропного препарата по следующим причинам: • Эти препараты достаточно липофильны. • Имеют большой объем распределения. • Их концентрация в тканях может превышать концентрации в плазме крови в 10-100 раз. Окончание действия однократной дозировки препарата для большинства пси-хотропных лекарственных веществ связано с процессом их перераспределения в организме. Примером может служить быстрое достижение седативного эффекта при внутривенном введении лоразепама, который быстро поступает из кровеносного русла в ткани головного мозга. В головной мозг поступает более значительная часть этой дозы, поскольку мик-роциркуляторное русло в ЦНС намного больше, чем в жировой ткани. Позже концентрация препарата в веществе мозга падает в связи с обратным поступлением лекарства из ЦНС в сыворотку крови и в дальнейшем в другие части тела. Именно это падение концентрации лежит в основе окончания непосредственного действия при однократном введении таких препаратов, как лоразепам. Понятие "распределение лекарственного вещества" означает процесс его накопления в различных частях тела (жировой клетчатке, костной ткани, мозговом веществе, жидкости и т.д.). Различия в скорости и уровне накопления лекарственного вещества в органах и тканях зависят от числа тех из них, в которых может происходить накопление. Даже в рамках одного компонента, каким является кровь, распределение может осуществляться среди нескольких субкомпонентов: • Собственно плазмы. • Белков плазмы крови. • Клеток крови (в частности, красные кровяные тельца). Большинство психотропных лекарственных веществ интенсивно связывается с белками [66-69]. Связанное с белками лекарственное вещество может составлять более 90% от общего содержания препарата в сыворотке крови. Величина свободной фракции препарата, хотя и очень незначительная в абсолютном исчислении, имеет огромное клиническое значение, так как именно она и определяет окончательное содержание вещества в месте его действия. Поэтому изменение в величине связанной фракции препарата с 95% до 90% может показаться и незначительным, но в отношении изменения величины свободной фракции это выражается в двукратном увеличении концентрации той части лекарственного вещества, которая собственно и оказывает действие в точке приложения препарата. Таким образом, любое изменение соотношения свободной и связанной фракций лекарственного вещества изменяет концентрацию лекарства в месте его действия и соответственно величину его эффекта. К причинам, вызывающим функциональное уменьшение белка в плазме крови, относятся: • Недоедание, например, при тяжелой анорек-сии. • Истощение, например, при нефротическом синдроме. • Пожилой возраст. • Сочетанное назначение лекарств, которые могут конкурентно связываться с белками [70,71]. Относительное увеличение свободной фракции лекарственно вещества может повысить его токсичность. Большинство лабораторных исследований, используемых при традиционном лекарственном мониторинге, не позволяет различать свободную и связанную фракции лекарственного вещества и соответственно определять эти динамические изменения. Для этого требуются особые, высокоспециализированные методики. Острые и хронические воспалительные заболевания повышают количество α1-кислого гликопротеина, который легко связывается с различными психотропными веществами. В этом случае происходит увеличение абсолютного количества связанного лекарственного вещества. При условии неизменной свободной фракции может создаться впечатление, что в крови находится чрезмерное количество вещества, но на самом деле происходит увеличение связанной (но биологически инертной) фракции. Абсолютная и относительная величины жировой ткани могут изменяться с возрастом или в связи с ожирением, вызванным болезненными причинами [31, 33,41,42]. Как указывалось выше, с возрастом понижается общее количество жидкости в организме и содержание белка, а процент жировой ткани повышается, что увеличивает резервуар для накопления психотропных веществ. Это объясняет, почему в пожилом возрасте психотропные препараты имеют более продолжительное действие. У больных, страдающих ожирением, также увеличивается это накопление и растягивается действие психотропных препаратов в зависимости от массы жировой ткани. По достижении состояния устойчивой концентрации формируются определенные пропорции между концентрацией препарата в плазме крови и его концентрацией в тканях [5, 72, 73]. На этом базируются методики лекарственного мониторинга, при которых определяется концентрация препарата в крови, что позволяет подтвердить адекватность дозировки лекарственного вещества и избежать возможного токсического действия. Психотропные препараты не оказывают своего действия, находясь в кровеносном русле, но их концентрация в крови уравновешивает концентрацию препарата в тканях. Хотя концентрация препарата в тканях может в 10-100 раз превышать концентрацию в плазме крови (в зависимости от конкретного органа), измерение последней концентрации позволяет косвенно судить о первой. МЕТАБОЛИЗМ Клинический пример: Больной, 62 года, на приеме у терапевта обнаруживает признаки депрессивного расстройства. В анамнезе отмечаются два инфаркта миокарда и связанная с ними сердечная недостаточность, проявления которой хорошо корригируются приемом дигоксина. В прошлом отмечается также злоупотребление алкоголем (последние 4 года алкоголь не употребляет). Врач назначил 75 мг амитриптилина на ночь per os. На повторном приеме, спустя неделю, у больного обнаруживается значительное усиление депрессивной симптоматики и врач увеличивает дозировку амитриптилина до 100 мг. Состояние больного продолжает ухудшаться, и его направляют к психиатру, который госпитализирует больного. При поступлении в больницу у больного отмечается подозрительность, настороженность, раздражительность и его госпитализируют с диагнозом психотической депрессии. Дополнительно ему назначают 10 мг галоперидола перорально на ночь. Пять дней спустя больной стал жаловаться на слабость и сердцебиения и вскоре был обнаружен у себя в палате без сознания. Его немедленно перевели в кардиологическую реанимацию, где при электрокардиографии была диагностирована неполная (2:1) атриовентри-кулярная блокада и периодически возникающие экстрасистолы. Лабораторный контроль общего уровня ТЦА в плазме крови показал концентрацию в 950 нг/мл. Все назначения психотропных препаратов были отменены. С падением содержания препаратов в крови устранилась кардиологическая и психотическая симптоматика. В данном случае у больного существовало несколько факторов риска, предопределивших развитие токсического эффекта ТЦА, несмотря на относительно низкую дозировку амитриптилина. Ниже мы переходим к обсуждению этих факторов. Процесс биотрансформации Большинство психотропных препаратов подвергается значительной биотрансформации с помощью процессов окисления, ведущих к образованию полярных метаболитов, которые затем выводятся с мочой. Обязательными этапами этой биотрансформации будет один или несколько из приведенных ниже типов: • Гидроксилирование • Деметилирование • Окисление. • Образование сульфоксидов. Большинство лекарственных препаратов до выведения подвергаются окислению в процессе биотрансформации (I фаза метаболизма), некоторые просто соединяются с такими частицами, как глюкуроновая кислота (II фаза метаболизма), а третьи выделяются без изменений (например, литий) [8,9,74]. II фаза метаболизма не зависит от функции печени, поскольку процесс конъюгации может происходить в большинстве органов. Таким образом, болезни печени не влияют на клиренс препаратов, которые проходят процесс глюкуронидизации. Это относится ко всем 3-гидроксибензодиазе-пинам (лоразепам, оксазепам, темазепам), которые при нормальной функции почек одинаково выводятся как в пожилом, так и в молодом возрасте. То же самое можно сказать и в отношении больных с выраженными нарушениями функции печени, что является основанием для применения 3-гидроксибензодиазепинов при раннем лечении алкогольного делирия. Если у таких больных и появятся признаки печеночной недостаточности, эти препараты по-прежнему будут без задержки выводиться из организма, не приводя к изменению когнитивных функций и психической активности. В противоположность этому для такого препарата, как диазепам, необходима обширная биотрансформация как обязательный элемент его выведения. Поэтому при печеночной недостаточности он может надолго задерживаться в организме [75]. Более того, метаболиты диазепама имеют сходные фармакологические свойства и, накапливаясь в организме, могут оказывать дополнительное фармакологическое действие. Биотрансформация путем окисления ведет к образованию метаболитов, чье фармакологическое действие может быть сходным или отличаться от действия изначального соединения. В любом случае действие этих метаболитов проявляется при формировании конечного фармакологического эффекта. Так, например, действие норфлуоксетина, по сути, не отличается от флуоксетина как в плане блокирования захвата серотонина, так и в плане ингибирова-ния некоторых CYP ферментов, но при этом данный метаболит выводится из организма намного медленнее [27, 28, 76]. В результате при длительном приеме флуоксетина происходит значительное накопление в организме норфлуоксетина, который в конечном счете определяет клинический эффект в большей степени, чем изначальное химическое соединение. Фармакологический профиль основного метаболита кломипрамина — дезметилкломи-прамина значительно отличается от профиля изначального соединения [77]. Тогда как кло-мипрамин является мощным ингибитором захвата серотонина, то дезметилкломипрамин — более мощный ингибитор захвата норадрена-лина. И поскольку значение кломипрамина при лечении обсессивно-компульсивных расстройств (ОКР) связано с его способностью блокировать захват серотонина, а не норадреналина, то это терапевтическое действие будет зависеть от соотношения между кломипрамином и дезметил-кломипрамином. Следовательно, если бы у больного процесс деметилирования кломипрамина происходил более интенсивно, то эффективность этого препарата при лечении ОКР была бы утрачена. Другая значимая ситуация возникает, когда в результате трансформации появляется метаболит, не имеющий столь же эффективного терапевтического действия, но при этом являющийся более токсичным соединением, чем изначальный препарат. Например, если бы уровень концентрации гидроксилированного метаболита имипрамина (2-гидроксиимипрамин) был более высоким, то этот трициклический антидепрессант был бы терапевтически малоэффективным и отличался бы высокой токсичностью [78]. Индуцирование CYP ферментов Алкоголь, никотин и большинство анти-конвульсантов индуцируют ряд CYP ферментов [25, 79-84]. Барбитураты и карбама-зепин по тому же механизму усиливают метаболизм других лекарственных веществ, также как и собственный (так называемая аутоиндук-ция). Достигнутый за первые 3-4 дня уровень концентрации названных препаратов в крови зависит в основном от скорости их выведения за это время. Однако в процессе дальнейшего приема препарата в результате аутоиндукции, которая сокращает период полувыведения, уровень концентрации существенно падает. Таким образом, по первичным данным мониторинга карбамазепина нельзя судить о возможной концентрации препарата по истечении нескольких недель приема, потому что его клиренс начинает возрастать в связи повышением ферментной активности (см. разд. "Альтернативные способы лечения" гл. 10). Действие алкоголя на скорость выведения лекарств, требующих обширной биотрансформации (таких как ТЦА), имеет трехфазный характер [64]. Однократный прием ТЦА и алкоголя с суицидальной целью у человека непьющего приведет к блокаде первичного метаболизма ТЦА. Это ингибирование повышает биодоступность ТЦА и может в три раза повысить пик их концентрации. Поэтому прием алкоголя на фоне высоких дозировок ТЦА может привести к летальному исходу. Регулярное употребление алкоголя в течение нескольких недель или месяцев может индуцировать CYP ферменты, что приведет к снижению уровня концентрации ТЦА. Таким образом, привычное, или субхроническое, потребление алкоголя может повышать активность печеночных ферментов и вызывать снижение уровня концентрации лекарственных веществ, выведение которых связано с обязательным окислением в процессе биотрансформации. Хроническое употребление алкоголя может привести к циррозу с уменьшением содержания CYP ферментов печени и массы печени, а также к портокавальному шунтированию. И если дозировка препарата не будет изменена с учетом снижения клиренса, то это будет проявляться повышением уровня концентрации лекарственного вещества в плазме крови. Ингибирование CYP ферментов В отличие от антиконвульсантов и алкоголя, такие лекарственные вещества, как квинидин, флекаинид, β-блокаторы, флуоксетин, пароксетин и антипсихотики, могут подавлять активность определенных CYP ферментов [27, 28,76,85-98]. Флуоксетин может ингибировать метаболизм трициклических антидепрессантов, отдельных бензодиазепинов, бупропиона, некоторых стероидных препаратов и антипсихотиков. Например, прием флуоксе-тина может вызвать увеличение концентрации такого ТЦА, как амитриптилин, в 2-10 раз, что делает обычные, ранее хорошо переносимые дозировки этого препарата весьма токсичными и даже угрожающими жизни [28, 29, 76].
|
|||||||||
|
|
Последнее изменение этой страницы: 2021-01-08; просмотров: 80; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.224.94.20 (0.051 с.) |

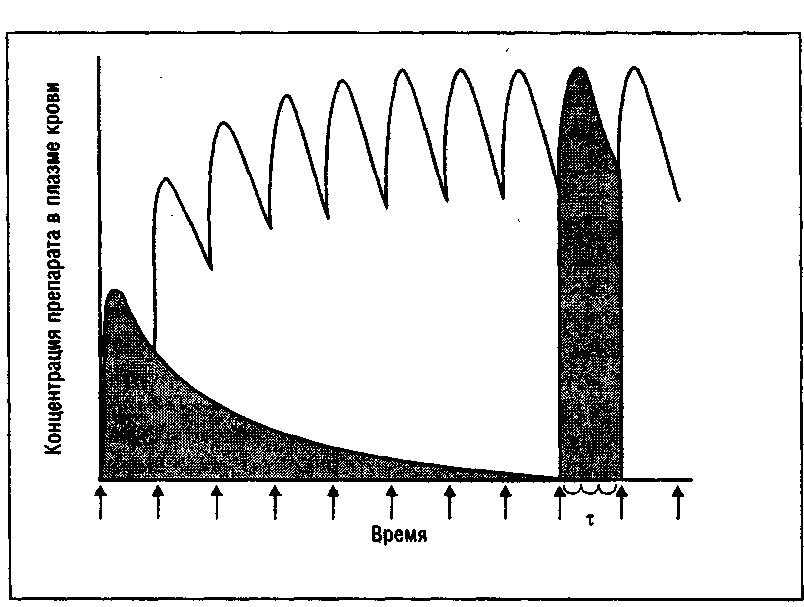 На рис. 3.3 показана фармакокинетика при однократной и при многократных дозировках лекарства, где по оси ординат отложены значения уровня концентрации препарата в сыворотке крови, а по оси абсцисс – отрезки времени между приемом назначений. При однократном пероральном приеме концентрация лекарственного вещества, достигнув Кmax затем относительно быстро понижается. Подобное первичное понижение концентрации связано в большей степени с процессом распределения лекарственного вещества, нежели с процессом выведения. Иными словами, первоначальное падение концентрации препарата связано не с выведением вещества из организма, а отражает скорость накопления вещества органами и тканями.
На рис. 3.3 показана фармакокинетика при однократной и при многократных дозировках лекарства, где по оси ординат отложены значения уровня концентрации препарата в сыворотке крови, а по оси абсцисс – отрезки времени между приемом назначений. При однократном пероральном приеме концентрация лекарственного вещества, достигнув Кmax затем относительно быстро понижается. Подобное первичное понижение концентрации связано в большей степени с процессом распределения лекарственного вещества, нежели с процессом выведения. Иными словами, первоначальное падение концентрации препарата связано не с выведением вещества из организма, а отражает скорость накопления вещества органами и тканями.


