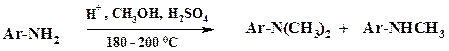Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Реагенты для проведения реакций алкилирования
Широко используются для проведения реакций алкилирования галогеналкилы. Некоторые из них получают галогенированием карбоновых кислот, кетонов, и альдегидов (см. раздел 2.1.6.7). Хлорметильные производные часто синтезируют с помощью реакций хлорметилирования (см. раздел 2.1.5.3). Однако наиболее часто галогеналкилы получают из соответствующих спиртов. Реагентами для замещения гидроксигруппы являются:
HCl, HBr, PCl3, PBr3, P + I2, SOCl2, PCl5, (POCl3)
Для синтеза хлоралкилов конц. HCl обычно не используют, т. к. выход целевого продукта из-за обратимости реакции невысок. Лучшие результаты получают с PCl3, SOCl2, но не POCl3, который в большинстве случаев дает эфиры фосфорной кислоты. Редко используют пентахлорид фосфора из-за его высокой стоимости и трудностей при загрузке в аппарат. В синтезе нембутала и барбамила изоамилбромиды получают из соответствующих спиртов с конц. HBr. Фосфортрибромид дает лучшие результаты, но данный реагент существенно дороже. Альтернативным методом синтеза этих промежуточных продуктов является взаимодействие изоамиларилсульфоната с KBr.
Ph-SO2Cl + i -HO-C5H11 → Ph-SO2-O-C5H11
Ph-SO2-O-C5H11 + KBr → i- Br-C5H11 + Ph-SO2-OH
Ph-SO2-OH + POCl3 → Ph-SO2Cl
Ph-SO2-OH + Ph-H +HO-SO2Cl → Ph-SO2Cl
Бензолсульфокислоту можно возвращать на стадию сульфохлорирования бензола. Выход изо -амилбромидов повышается на 14 %. Однако внедрение этого способа требует значительных капитальных вложений т. к. необходимо создавать участок по утилизации бензолсульфокислоты. Алкилиодиды более реакционноспособны, но их стоимость намного превышает стоимость алкилбромидов и алкилхлоридов. В связи с этим в промышленности алкилиодиды обычно не используют. Кроме алкилгалогенидов в качестве реагентов при получении алкиламинов применяют более дешевые спирты при кислотном катализе.
Alk-OH + H+ → Alk(OH2)+ → Alk+ + H2O
Если реакционной способности метил- или этилоксониевых соединений недостаточно, используют диметил- и диэтилсульфаты. Для введения более длинной алкильной цепочки могут быть использованы эфиры бензолсульфокислоты, но их реакционная способность невелика. Поэтому чаще всего используют алкилбромиды. Для проведения реакций гидроксиэтилирования наиболее дешевым и безопасным реагентом является этиленхлоргидрин (HO-CH2-CH2-Cl). Введение гидроксиалкильных остатков осуществляют также и с помощью оксида этилена и его производных (производных оксирана). Но оксиды этилена и пропилена высокопожаро- и взрывоопасны. Работу с ними обычно проводят непосредственно на предприятиях, которые их и выпускают, без транспортировки баллонов с ними. Более безопасным реагентом является 3-хлор-1,2-эпоксипропан (эпихлоргидрин). Другие производные оксиранов получают непосредственно в синтезе препаратов, например при получении фенилэфрина.
Механизмы реакций
В зависимости от заместителей, которые находятся у атома углерода, связанного с нуклеофугной группой, а также и от растворителя, реакции могут идти по механизму асинхронного (SN1) и синхронного замещения (SN2).
Асинхронный механизм реакций. В 1925 г. Лоури предположил, что первоначально происходит диссоциация связи С-Х с образованием карбокатиона, который впоследствии реагирует с нуклеофилом:
Элементарные стадии процесса диссоциации представлены на следующей схеме:
Через энергетический барьер соединение превращается в переходное состояние, в котором связь С-Х растянута. Затем происходит перенос электрона к более электроотрицательному атому Х (тесная ионная пара), на этой стадии заметную роль играет энергия сольватации образующихся ионных зарядов. Для реализации данного механизма очень важна роль растворителя. Полярные протонные растворители в большей степени, чем неполярные или апротонные способны увеличивать расстояние между сольватированными ионами, благодаря чему суммарная свободная энергия системы уменьшается.
Закономерности. При кинетическом исследовании реакций найдено, что скорость реакции (в начале процесса) зависит только от концентрации электрофила. Лимитирующей стадией является разрыв связи С-Х, а взаимодействие иона карбония с нуклеофилом протекает быстро. Изучение реакции дифенилхлорметана (бензгидрилхлорида) с нуклеофилами в жидком SO2 с фторид ионом и пиридином дает одну и ту же константу скорости реакции и не зависит от концентрации нуклеофилов. Таким образом, можно записать следующее кинетическое уравнение:
V = k [R-X]
Для гидролиза бутилхлорида найдены величины DH¹ = 82 кДж/моль и DS¹ = + 12 кал. град-1 .моль-1 (э. е.). Величина энтропии активации показывает на образование пары зарядов в переходном состоянии. Корреляционный анализ реакций по уравнению Тафта показал, что наблюдается неочевидная закономерность – величина коэффициента реакционной серии r = -3…- 5.Следовательно, электронодонорные заместители ускоряют реакцию. Это объясняется тем, что они облегчают делокализацию положительного заряда и тем самым стабилизируют карбокатион. Структура карбокатиона является плоской, поэтому нуклеофил равновероятно может атаковать как сверху, так и снизу плоскости, как это проиллюстрировано на схеме:
В условиях протекания процесса SN1 хиральные соединения дают рацемическую смесь. В тех случаях, когда отсутствует возможность рацемизации продукта и практически нет побочных реакций, на механизм процесса можно не обращать внимание. Однако в синтезе БАВ требуется высокая стереоселективность и чистота получаемых продуктов. Реакция часто сопровождается протеканием побочных процессов. В карбокатионе возможно перемещение заряда на соседний атом углерода, тогда образуется смесь соединений. Кроме того, карбокатион может стабилизироваться за счет отщепления протона и при этом образуется олефин. Последний процесс (реакции дегидрогалогенирования) целенаправленно используется в органическом синтезе.
Рассмотренные закономерности свидетельствуют о том, что необходимо избегать при получении ЛС протекание реакций нуклеофильного замещения у насыщенного атома углерода по механизму SN1.
Синхронный (бимолекулярный) механизм реакций. В 1929 г. Лондон сформулировал основные положения бимолекулярного механизма реакции.
В переходном (симметричном) состоянии молекулы связи C-X и C-Y имеют длину больше, чем ковалентные связи, и являются частично ионными. В связи с тем, что атака нуклеофила идет с тыла связи С-Х (нуклеофил «садится на хвост» s-орбитали С-Х), в случае исходного хирального соединения полученный продукт является также хиральным, но знак вращения плоскости поляризованного света меняется на противоположный. Это так называемое Вальденовское обращение. Когда бы ни происходило изменение зарядов на Х и Y – в самом начале, в переходном состоянии или конечной стадии замещения – огромную роль имеет энергия сольватации. Так в реакции иодистого метила с хлорид ионом при переходе от метанола (протонный полярный растворитель) к диметилформамиду (апротонный полярный растворитель) скорость увеличивается в 1,2×106 раз. CH3-I + Cl - → CH3-Cl + I -
В реакциях, протекающих по механизму SN2, в отличие от SN1, используют апротонные растворители (см. раздел 1.4, табл.1.4).
Закономерности. Бимолекулярное замещение при малых концентрациях реагентов описывается кинетическим уравнением второго порядка:
V = k 2 [R-X] [Y -]
Энтропия активации составляет величину - 20 …- 28 э. е., т. е. переходное состояние более упорядоченное и компактное по сравнению с исходной системой. Однако при большом избытке нуклеофила процесс описывается кинетическим уравнением первого порядка, т. к. порядок по нуклеофилу нулевой:
V = k 1 [R-X]
Скорость реакции зависит от заместителей, находящихся у атома углерода, нуклеофугной группы, растворителя и нуклеофила. Влияние заместителей: при замещении брома на этоксильную группу в абсолютном спирте при 55 оС относительная скорость реакции уменьшается при увеличении длины алкильного радикала.
R-Br + EtO- → R-O-Et: R (k относит): CH3 – 17,6; C2H5 – 1; н -C3H7 – 0,31; н -C4H9 – 0,23; н -C5H11 – 0,21
При разветвлении углеродного остатка (реакция гидролиза алкилбромидов в 80 % спирте при 25 оС) наблюдались следующие константы скорости реакции: CH3Br – 2 140 л.моль-1 с-1, Et-Br – 170 л.моль-1 с-1, для изопропилбромида константа скорости второго порядка равна 4,7 л.моль-1 с-1, но одновременно идет и мономолекулярная реакция с константой скорости 0,24 с-1. Для третбутилбромида в этих условиях идет только мономолекулярная реакция, константа скорости 1,01 . 10-3 с-1. Реакции бимолекулярного замещения относятся к процессам, идущим при орбитальном контроле: скорость реакции зависит от величины коэффициента АОНСМО на атоме углерода электрофила и в целом от энергии НСМО. Чем ниже эта энергия, тем легче идет реакция. Электроноакцепторные заместители понижают, а электронодонорные повышают энергию НСМО. Скорость процесса зависит также и от характера нуклеофугной группы. Так для производных, в зависимости от характера группы Х, в соединении R-X скорость увеличивается в следующем ряду: F < Cl ≤ Ar-SO2-O < Br < I. В реакциях бимолекулярного замещения электрофилы являются мягкими реагентами. Со стороны нуклеофила реакционная способность может быть описана уравнением Свена-Скотта, аналогичным уравнению Гаммета:
lg k / k 0 = s n
где s – постоянная, характеризующая реакционную серию (аналогична r в уравнении Гаммета), n – сила нуклеофила (аналогична s в том же уравнении). Уравнение Свена-Скотта разработано на примере реакции гидролиза бромистого метила водой (для Н2О n = 1). Величины n приведены для широкого ряда нуклеофилов, реагент (n): H2O (0), F- (2), CH3COO- (2,7), пиридин (3,6), Cl- (3,04), Br- (3,9), N3- (4,0), HO- (4,24), C6H5-NH2 (4,5), NH3- (4,8), H-N(CH3)2 (5,0), CN- (5,1), I- (5,2), C2H5O- (5,86), SH- (6,5). Как видно, мягкие реагенты обладают большей нуклеофильностью. При изменении механизма реакции нуклеофильность реагентов меняется, в случае реакций SN1 наиболее сильным нуклеофилом является гидроксид анион.
Алкилирование аминов
Наиболее многотоннажным производством является синтез алкиламинов. Так при алкилировании аммиака метанолом в присутствии дегидратирующих катализаторов (Al2O3, W2O5) в парах при температуре 350 оС и давлении 0,6 МПа получают смесь метиламина, диметиламина и триметиламина. В колонне при давлении 0,4 МПа отгоняют амины и аммиак, которые сжимают компрессором до 1,8 МПа и разделяют на ректификационной колонне. Триметиламин вновь направляют на первую стадию, где это соединение вступает в реакцию с аммиаком, давая как метиламин, так и диметиламин. Аналогично получают этиламин, диэтиламин и триэтиламин. Хотя аппаратурное оформление процесса довольно сложно, использование дешевых спиртов более рентабельно, чем диметилсульфата или диэтилсульфата.
Также высок тоннаж производства N- алкиланилинов:
Реакцию ведут в каскаде автоклавов непрерывным методом в присутствии каталитических количеств серной или фосфорной кислоты. Так же, как и при получении алкиламинов, образуется смесь моно- и диметиланилинов. Кислота в первую очередь связывается с аминогруппой анилина. Однако частично наблюдается и протонирование метанола с последующим разложением до метилкатиона и воды. Аналогично осуществляют этилирование анилина.
Реакцию ведут в каскаде из трех реакторов, температура в первом из них 200 оС, а в последнем 280 оС. В качестве катализатора используют PCl3. Полученную смесь направляют на ректификацию. Еще одним из крупнотоннажных производств является синтез этаноламинов:
Смесь этаноламинов разделяют ректификацией. Моноэтаноламин и диэтаноламин применяют в синтезе лекарственных препаратов. Триэтаноламин хорошо растворяет сероводород и может быть использован для его улавливания. Заменой гидроксильных групп на хлор из триэтаноламина получают азотиприт. Взаимодействие этиленоксида с соляной кислотой дает этиленхлоргидрин, который широко используют в синтезе синтетических лекарственных средств.
Для введения диалкиламиноэтиленовой цепочки обычно этиленхлоргидрин превращают в N- диалкиламиноэтаноламин, а затем гидроксильную группу замещают на хлор. Полученный продукт является алкилирующим средством.
Для синтеза метильных производных применяют алкилирование диметилсульфатом, а этильных – диэтилсульфатом, которые являются промышленно производимыми продуктами. Как алкилирующее средство диэтилсульфат менее реакционноспособен, чем диметилсульфат. Поэтому этилирование малореакционноспособных веществ осуществляют этилбромидом. Алкилирование алкилгалогенидами обычно ведут в апротонных растворителях с использованием кислотосвязывающих средств: карбонат-, бикарбонат натрия, триэтиламин или пиридин. Если реагирующий амин дешев, то он может служить кислотосвязывающим средством. Как уже неоднократно отмечалось, обычно при алкилировании аминов образуется смесь продуктов. В синтезе лекарственных средств необходимо получение веществ высокой чистоты. Разделение смесей редко дает хорошие результаты, к тому же удорожает производство. Наименьшие проблемы возникают при синтезе третичных аминов. Для получения первичных и вторичных аминов имеются специальные приемы.
|
|||||||||
|
|
Последнее изменение этой страницы: 2016-04-08; просмотров: 603; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.225.35.81 (0.031 с.) |