
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Віроїд сонячного опіку авокадо.
Являють собою поліпротеіди з невеликою масою (20-30D) (рис. 2). Особливістю інфекційного процесу є відсутність асоційованих з пріонами нуклеїнових кислот. Мабуть, на відміну від інфекційних захворювань бактерійного або вірусного походження, у разі т.н. неканонічних вірусних інфекцій саме білки є безпосередніми збудниками захворювання і саме вони виявляються в уражених органах (передусім в ЦНС) у великих кількостях. Рис. 9. Білки - пріони
Згідно з гіпотезою лауреата Нобелівської премії 1997 року Стенлі Прузінера механізм запуску процесу патогенезу у разі неканонічних інфекцій пов'язаний з тим, що екзогенний білок (пріон) потрапляючи в клітину змінює конформацію клітинних білків (які в нормі вбудовуються в мембрану клітини) відповідно до власної структури і накопичуються в клітині у високих концентраціях, викликаючи безповоротні зміни кліток нервової тканини. Відрізняючись досить високою стійкістю до термічної обробки пріони можуть ініціювати патологічні процеси при вживанні м'ясних продуктів, що не пройшли достатньої обробки. Не можна виключити також гіпотезу про те, що пріони можуть бути продуктами дерепресії яких-небудь “сплячих" клітинних генів, що блокують нормальне функціювання клітинного геному.
Фізико-хімічні властивості пріоновых білків: ü ультромікроскопічні розміри (здатні проникати через бактеріальні фільтри); ü стійкі до великих доз УФ опромінення; ü стійкі до нагрівання при 80оС (неповна інактивація при 100оС); ü стійкт до 0,1 – 0,5 мМ NH2OH; ü повна інактивація при автоклавуванні при 136оС протягом 18 хвилин; ü інактивація сухим жаром 160оС протягом 24 годин;
ü інактивуються 1N розчином питноїсоди протягом 1 години при 20оС; ü інактивуються 100% фенолом; ü інактивуються 3 - 8 М розчином мочевини; ü інактивується 1 – 10% SDS; ü інактивуються 2,5% розчином гіпохлорида натрію. Біологічні властивості пріонових білків: ü відсутність экліпс-фази; ü тривалий інкубаційний період (від місяців до десятків років); ü хронічна прогресуюча патологія (повільна інфекція); ü відсутність запальних реакцій; ü відсутність ремісій і одужання: неминуче летальна інфекція; ü відсутність продукції інтерферону; ü нечутливість до інтерферону; ü відсутність антигенності, відсутність білків, відмінних від білків хазяїна; ü не викликають імунної відповіді (як гуморального так і клітинного) ураженого організму; ü відсутність патогенної дії на клітини in vitro; ü різні чутливість, вірулентність і патогенез (ураження різних зон мозку у різних видів); ü зміна круга хазяїв при пасивуванні; ü реплікація до титрів 105-1011 в мозку; ü відтворення як в клітині, так і в штучної, безклітинної, системі ü автокаталітичним шляхом
Особливості патогенезу: Спочатку репликується в селезінці і інших органах ретикуло-ендотеліальної системи, а потім в мозку; адаптується до нового хазяїна (скорочення інкубаційного періоду); присутня генетична регуляція чутливості у деяких видів (у овець і мишей для збудника скрейпи); специфічний круг хазяїв у кожного штаму; наявність штамів з різним кругом хазяїв і вірулентністю. Особливістю іфекційного процесу, викликаного пріонами являється відсутність асоційованихз пріонами нуклеїнових кислот. Мабуть, на відміну від інфекційних захворювань бактеріального або вірусного походження, у випадку т.з. неканонічних вірусных інфекцій саме білки є безпосередніми збудниками захворювання і саме вони виявляються в уражених органах (передусім в ЦНС) у великих кількостях.
Рис. 10. Зріз кори головного мозку при губчастій енцефалопатії. Зліва – нормальна тканина, справа – ураження мозку при губчастій енцефалопатії
Звідси і назва: "губчаста хвороба мозку". Відрізняючись досить високою стійкістю до температурних дій, пріони можуть ініціювати патологічні процеси при вживанні м'ясних продуктів, що не пройшли достатню обробку. Окрім вступу ззовні (наприклад, з їжею, переливанням крові і її продуктів, вступом гормональних препаратів), пріонова інфекція може бути "запущена" мутацією гена, що кодує в нормі синтез одного з мембранних білків, - PrP - c. Нормальний клітинний білок - PrP - c - кодується єдиним геном, розташованим у людини в 20 хромосомі. Складається приблизно з 254 амінокислотних залишків, включаючи 22-членний N -терминальный сигнальний пептид. Нормальна форма є мембранним білком нервових клітин, схильним до швидкого виведення і протеолітичного розщеплення на поверхні клітин. Мутантна ізоформа пріонового білка PrP-c - інфекційний пріоновий білок PrP - Sc має молекулярну масу, аналогічну нормальній вазі пріон-протеїна і кодується тим же геном. Обидві форми стійкі до дії протеаз. Спектроскопічний аналіз показав, що PrP-c і PrP-Sc мають різну конформацію: в PrP - c домінує α-спіралізація і в дуже малих кількостях виявляються β-складчасті структури (3%), тоді як PrP - Sc мають високий вміст β-структур (до 43%) (рис. 11)
Малиновий колір – α -спиралі; зелений – β –складки.
Патогенна форма пріона має здатність дифундувати крізь клітинну мембрану і накопичуватися як усередині нервових клітин, так і поза ними. Патогенні форми пріонів властива здатність до утворення міжмолекулярних асоціатів, що обумовлюють, зокрема, утворення амілоїдів в уражених нервових тканинах. PrP - c входить до складу зовнішніх клітинних мембран, пов'язаний із зовнішньою поверхнею клітин якорем гліколіпіду і бере участь в эндоцитозі і катаболізмі клітин. У нейронах пріон-протеїн потрібний для нормальної синаптичної функції. Передбачається, що в нормі пріони беруть участь в міжклітинному пізнаванні і клітинній активації. Не дивлячись на те, що найвищий рівень концентрації PrP - c виявлений в нейронах, його можуть синтезувати і багато інших клітин організму. Патогенні пріон-протеїни, здатні до трансмісії, є мутантами клітинної ізоформи нормального пріон-протеїна. До 1991 року встановлено (Prusiner S.B.) 18 різних мутацій гена PrP людини, які пов'язані з різними пріоновими хворобами. PrP - c здійснює в клітині циклічне пересування. Він синтезується в шорсткому ендоплазматичному ретикулуме, потрапляє в апарат Гольджи і переноситься за допомогою екзоцитозу на поверхню клітини прикріпляючись до клітинної мембрани за допомогою глікозил-фосфатідил-інозитольного (ГФІ) хвоста. Після цього він переноситься в клітину за допомогою эндоцитоза, а потім назад на поверхню. Цей цикл здійснюється за 60 хвилин. При цьому PrP - c може розрізати в двох ділянках приблизно в середині молекули, а також на рівні ГФІ хвоста. Це може відбуватися, як в эндоцитозном бульбашці, так і на поверхні клітини. Одна із спроб пояснити механізм патогенезу пріонових інфекцій (Мотузок С. В., 1999) зв'язує патогенність пріонових білків з їх здатністю до проходження через клітинні мембрани і зв'язування з протеїназами. У результаті патогенний пріон перетворюється на транспортний білок, що доставляє протеїнази в міжклітинний простір. Наслідком цих подій, відповідно до гіпотези С. В. Вірьовки, являється фізіологічно недоречна активація внутрішньоклітинних проферментів з наступним розвитком нейродегенеративных процесів, що ведуть до загибелі клітини.
Вважається що усі встановлені до теперішнього часу види трансмісивних спонгіоформних енцефалопатій людини і тварин пов'язані між собою єдиним збудником і розрізняються лише за механізмом передачі інфекціі та за реакцією макроорганізма на інфекційний агент. Білок PrP Sc, так само як і усе інші прионы утворює агрегати у виді високо упорядоченых волокнини, що також називається амілоїдами. Ця властивість украй зближує його з білками інших нейродегенеративных захворювань (амілоідозів), таких як хвороби Альцгеймера і Хантигтона. Для хвороби Альцгеймера характерне накопичення агрегатів білку beta -амилоида в мозку. При хворобі Хантингтона відбувається ампліфікація триплета, що кодує глутамін, в гені білка Хантингтина і ця мутація викликає агрегацію білку. Функціональна роль білка PrP доки не відома, проте нещодавно було показано, що PrPC може зв'язуватися з іонами міді. Передбачається що такий комплекс, за допомогою эндоцитоза потрапляючи в клітину потім диссоціює і іон міді переноситься з эндоцитозного бульбашки всередину клітини в цитоплазму.
|
|||||||
|
|
Последнее изменение этой страницы: 2017-02-17; просмотров: 179; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.119.133.228 (0.01 с.) |
 1.6. Пріони - як і деякі віруси є збудниками повільних летальних інфекцій, що викликають поразки центральної нервової системи людини і тварин, об'єднаних в групу “підгострих трансмісивних губкоподібних енцефалопатій". Включають чотири хвороби людини (куру, Крейцфельда-Якоба, синдром Герстманна-Штрейснера, аміотрофічний лейкоспондіоз) і чотири захворювання тварин (скрейпі овець, губкоподібну енцефалопатію корів (“сказ" корів), трансмісивну енцефалопатію норок і хронічну виснажуючу хворобу лосів, що знаходяться в неволі, чорнохвостого оленя).
1.6. Пріони - як і деякі віруси є збудниками повільних летальних інфекцій, що викликають поразки центральної нервової системи людини і тварин, об'єднаних в групу “підгострих трансмісивних губкоподібних енцефалопатій". Включають чотири хвороби людини (куру, Крейцфельда-Якоба, синдром Герстманна-Штрейснера, аміотрофічний лейкоспондіоз) і чотири захворювання тварин (скрейпі овець, губкоподібну енцефалопатію корів (“сказ" корів), трансмісивну енцефалопатію норок і хронічну виснажуючу хворобу лосів, що знаходяться в неволі, чорнохвостого оленя).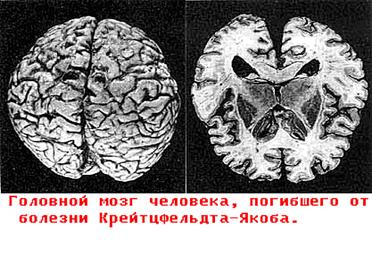 Відповідно до гіпотези лауреата Нобелівської премії 1997 року Стенлі Прузинера механізм патогенезу, пов'язаний з формуванням в корі головного мозга структур, на крезові тих, що нагадують губку(рис. 8).
Відповідно до гіпотези лауреата Нобелівської премії 1997 року Стенлі Прузинера механізм патогенезу, пов'язаний з формуванням в корі головного мозга структур, на крезові тих, що нагадують губку(рис. 8). Рис.11. Просторова структура нормального (ліворуч) і пріонового (справа) білків.
Рис.11. Просторова структура нормального (ліворуч) і пріонового (справа) білків.


