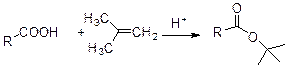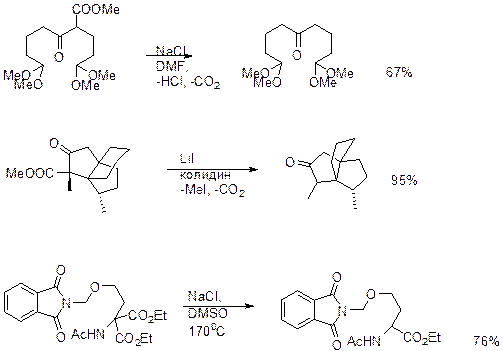Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Третбутилдиметилсилиловые эфиры TBSСтр 1 из 5Следующая ⇒
Курс Лекций «Современные проблемы органической химии»
Часто требуется в процессе синтеза защита и активация функциональных групп.
В процессе органического синтеза с использованием полифункциональных соединений очень часто возникает проблема защиты одной из них с целью сохранить в процессе превращений. Затем после проведения ряда последовательных превращений уже в конечном продукте снять эту защиту – регенерировать функциональную группу и использовать ее в дальнейших превращениях.
Эти защитные группы должны удовлетворять следующим условиям:
1. Быть дешевыми и легко доступными 2. Должны легко и эффективно вводиться 3. Должны легко характеризоваться и избегать таких сложностей как создание новых стереогенных центров. 4. Должны быть стабильными в реакционных условиях и при обработке 5. Должны быть устойчивы в условиях разделения, очистки и хроматографирования 6. Должны легко удаляться селективно и полностью при весьма специфических условиях. 7. Побочные продукты снятия защитных групп должны легко отделяться от субстрата
Чаще всего в процессе синтеза приходится защищать гидроксильную, диольную, карбоксильную и аминную группы – т.е. наиболее важные группировки природных веществ и их синтетических аналогов. Эти группировки или их производные наиболее часто встречаются в лекарственных соединениях.
При этом каждая из защитных групп должна отщепляться такими реагентами и в таких условиях, в которых не затрагивались бы другие защитные группы.
Давайте вначале рассмотрим способы удаления и отщепления защитных групп.
1. Самый простой способ это основной сольволиз.
Ацильная защита широко используется для защиты SH, OH и аминогрупп. Ацильная защита легко снимается в условиях щелочного гидролиза. Эфиры тиолов очень чувствительны к нуклеофильной атаке, поэтому для защиты используются ацетаты, бензоаты и пивалаты. Ацетаты и бензоаты особенно ценимы, т.к. легко отделяются K2CO3 или NH3 в MeOH. Стерические препятствия ацильного радикала важны как и электронные эффекты в α положении к карбонилу. Пивалаты из-за стерических препятствий значительно медленнее могут быть удалены селективно. Трифторацетаты очень легко гидролизуются при pH 7.
Влияние электронных эффектов заместителей демонстрирует следующий ряд:
Конечно, когда имеем дело с защищенными сложноэфирными группами полифункциональными соединениями здесь могут возникать проблемы внутримолекулярной переэтерификации, связанной с миграцией ацильных групп на соседние спиртовые. Эта проблема более существенна для бензоатов, чем для ацетатов.
Амиды. Трифторацетамиды очень лабильны и легко удаляются с K2CO3 в MeOH – аналогично метиловым эфирам.
Если мы перейдем к фталимидам, то их расщепление гидразином в MeOH или EtOH - не строго реакция сольволиза. На первой стадии имеет место атака гидразина на карбонильную группу имида (более чувствительна к атаке нуклеофильной, чем в амиде).
2. Защитные группы, удаляющиеся с помощью кислот.
Такое расщепление имеет место в случае третичных бензиловых и алкиловых эфиров.
Реакция требует промотирования H+ или кислот Льюиса. Здесь широкий разброс условий: например HBr в AcOH снимает защиту с бензиловых эфиров, а третиловые (Ph3C-) эфиры уже расщепляются разбавленной уксусной кислотой.
Вторая защитная группа, которая легко снимается в условиях кислого гидролиза – это ацетальная защита. В качестве кислот используют H+ или кислоты Льюиса.
CH3-CH(OMe)2
3. Расщепление тяжелыми металлами.
Расщепление О,О ацеталей, O,S и S,S- ацеталей в условиях гидролиза можно катализировать солями Ag (I) и Hg (II).
4. Расщепление связей Si-C и Si-O под действием фтораниона.
Силильная защита - одна из наиболее перспективных защитных групп, которая разрабатывается в последние 20 лет. R3Si группа силиловых эфиров легко удаляется в условиях кислого и щелочного гидролиза.
Однако триалкилсилильная группа может быть снята действием Bu4NF в ТГФ или HF в ацетонитриле, KF в ДМСО. Это обусловлено возможностью взаимодействия F- с вакантными орбиталями атома Si с образованием пентакоординационного атома кремния.
В качестве защитной группы может выступать триметилсилилэтильная группа, которая легко снимается действием F-.
В основе всех этих процессов лежит реакция β-элиминирования.
5. Снятие защитных групп с помощью цинка.
Этот тип снятия защитной группы можно продемонстрировать следующим примером трихлорэтилового эфира.
6. Снятие защиты с использованием реакции β-элиминирования.
Пример снятия защиты 9-флуоренилметоксикарбонильной (Fmoc). Механизм E1CB. Пиперидин или морфолин в ДМФА.
Fmoc защита используется в пепетидном и гликопептидном синтезе.
7. Снятие защиты реакцией гидрогенолиза.
Этот метод годен для снятия бензильной защиты в простых и сложных эфирах, карбоматах и аминах. Связь O-CH2Ph и N-CH2Ph легко расщепляется в условиях гидрогенолиза в присутствии Pd и др. переходных металлов. Иногда используют Ni Ренея. Бензильная защита используется в основном в синтезе пептидов.
8. Удаление защитной группы окислением.
Этот метод используется в синтезе п -метокси- и 3,4-диметоксибензиловых эфиров. В качестве окислителя используют 2,3-дихлор-5,6-дициано-1,4-бензохинон (DDQ). Идет перенос e- к DDQ.
9. Снятие защиты с помощью металлов в растворах.
Na или Li в жидком аммиаке расщепляет бензиловые простые и сложные эфиры в присутствии источника протонов с отщепленной алкоксидной или карбоксилатной уходящих групп.
10. Нуклеофильное расщепление связи C-O.
Это классическое нуклеофильное замещение. Нуклеофилы хлорид, иодид, цианид, тиофенолят в биполярном апротонном растворителе при повышенной температуре.
Ограничивается метиловыми и этиловыми эфирами. Большую роль играет стабилизация карбоксилат аниона, чем она выше, тем реакция идет легче.
Аналогично реагируют и фениловые эфиры.
11. Аллильные защитные группы.
Аллильные группы требуют для своего снятия мягких и специфических условий не затрагивающих других функциональных и защитных групп. Аллильная группа не используется для защиты спиртов, карбоновых кислот (аллиловые эфиры) и аминов (аллилкарбаматы).
Аллиловые простые эфиры стабильны в сильно щелочной среде, но расщепляются t BuOK в ДМСО, включая двухступенчатый процесс, вовлекающий изомеризацию двойной связи. При этом образуется эфиры енолов, которые гидролизуются мягкими кислотами.
Для целей снятия защиты можно использовать комплексы Rh (I). Возможный химизм процесса показан далее.
12. Снятие защиты при действии УФ.
о-Ниторбензиловые эфиры (спирт), о-ниторбензиловые карбаматы (амин), о-ниторбензилкарбонаты (спирт), о-ниторбензиловые сложные эфиры (карбоновые кислоты), о-ниторбензилиденацетали (1,2- и 1,3-диолы).
Схема протекания процесса.
13. Снятие защитных групп.
Это случай когда защитная группа устойчива в широком интервале реакционных условий и в процессе реакции трансформируется в новую группу, которая лабильна и легко отщепляется в мягких условиях.
14. Временные защитные группы.
Когда требуется защитить на 1-2 стадии. Для того, чтобы провести реакции по менее активной кетонной функции оксоальдегида А. Альдегидную функцию защищают N-метиланилидом лития, затем проводят реакцию по кетогруппе. При водной обработке реакционной смеси временная защитная группа симается.
Еще один пример силилирования 3-формилфурана.
Защита гидроксильной группы
Гидроксильная группа может быть защищена с помощью:
а. ацилирования – сложные эфиры; б. силилирования – силиловые эфиры.
Силиловые эфиры SiMe3, SiEt3, t- BuMe2Si, t- BuPh2Si, i- Pr3Si – различие в радикалах изменяет стерический объем Si-радикала и повышает избирательность силилирования.
в. алкилирования – метиловые, бензиловые, п -метоксибензиловые и 3,4-диметоксибензиловые, тритиловые, третбутиловые, аллиловые, аллилоксикарбонильные производные. г. алкоксиалкильные эфиры – ацетали, метоксиметиловые (МОМ MeOCH2), метилтиометиловые (MeSCH2), (2-метоксиэтокси)метиловый (MeOCH2CH2OCH2), бензилоксиметиловый (ВОМ С6H5CH2OCH2), β -(триметилсилил)этоксиметиловый эфир (Me2SiCH2CH2OCH2O).
Сложные эфиры
Для этерификации ангидриды, галогенангидриды кислот в пиридине. О снятии защитной ацильной группы мы уже говорили. В основном это щелочной гидролиз и сольволиз, на скорость которых сильное влияние оказывает стерическое препятствие.
NH3 в МеОН t- BuCO < PhCO < MeCO < ClCH2CO CF3CO отщепляется уже при рН 7 Следует учитывать, что в
Пивалоильная группа для снятия требует жестких условий (КОН в МеОН), которые могут снимать другие защитные группы, например, силильные. Решение этой проблемы иллюстрирует следующий пример:
Часто для снятия ацильной защиты используют энзимы, эстеразы, липазы, и тогда можно получать энантиомеры. Следующий пример показывает использование липаз для селективной защиты и гидролиза эфиров на примере Castanospermine – ингибитора человеческого иммунодефицитного вируса.
Этерификация по С1 в присутствии субтилизина, затем используя липазу получают 1,7-дибутаноилпроизводное.
Как уже говорилось, ацилирование традиционно – ангидриды и галогенангидриды в пиридине 0-200С. Третичные спирты ацилируются очень медленно и реакцию катализируют 4-диметиламинопиридином (каталитическое количество). Пивалоилхлорид осуществляет селективное ацилирование первичной гидроксигруппы в присутствии вторичных.
Силиловые эфиры
Используют достаточно активно с 1970 года. Силильные защитные группы очень популярны. Легко вводятся, отщепляются в мягких условиях и стабильны в условиях многих реакций. Варьируя заместители у атома Si, можно изменять стерический объем и за счет электронных эффектов изменять легкость элиминирования.
Si-защитные группы с объемными заместителями более стабильны в условиях кислого и щелочного гидролиза, действию литий- и магнийорганических соединений, окислению, восстановлению и в условиях колоночной хроматографии., чем с радикалами небольшого объема.
Введение фенильных групп к Si увеличивает стабильность этой группы в условиях кислого гидролиза. Ph3Si/Me3Si = 1:400.
В щелочных условиях стабильность почти одинакова. Ph3Si/Me3Si = 1 (электроотрицательные группы – дефицит на Si).
По стабильности к элиминированию в кислых условиях триорганосилильные группы можно расположить в ряд: Me3Si(1) < Et3Si(64) < t- BuMe2Si(20’000) < i- Pr3Si(700’000) < t- BuPh2Si(5’000’000)
Стабильность в условиях щелочного гидролиза:
Me3Si(1) < Et3Si(10-100) < t- BuМe2Si = t- BuPh2Si(20’000) < i- Pr3Si(100’000) Триметилсилиловые эфиры TMS
Me3SiO группа отщепляется в очень мягких условиях АсОН или К2СО3 в МеОН. Скорость гидролиза зависит как от стерических, так и от электронных эффектов. Увеличение стирики уменьшает скорость гидролиза. Электроноакцепторные функции в алкогольной части увеличивают скорость.
Получение. Для триметилсилилирования используют большое количество реагентов, большинство из которых являются коммерческими.
Дешевый Me3SiCl (TMSCl) и более активный CF3SO2OSiMe3 (TMSOTf) силилируют гидроксильные группы в присутствии оснований (пиридин, Et3N, i- Pr2NЕt, имидазол). Соли аминов гидрохлориды отфильтровывают; для трифлата нужна водная обработка. В качестве растворителей могут быть использованы CH2Cl2, MeCN, ТГФ, ДМФА (абсолютные).
В случае альдегидов и кетонов TMSOTf превращает их в силиловые эфиры енолов.
Трифлат может расщеплять эпоксиды.
Когда присутствие солей или водная обработка нежелательны триметилсилилирование может быть осуществлено: N,O-бис(триметилсилилацетамид) катализируемый Me3SiCl, 1-триметилсилилимидазол (TMSIM).
Триэтилсилиловые эфиры TES
Et3Si группа более стабильна, чем метильная при колоночной хроматографии и к действию окислителей, восстановителей и металлоорганических соединений.
TES в 10-100 раз более стабильна к гидролизу и нуклеофильной атаке, чем Me3Si, но менее стабильна, чем t- BuMe2Si (TBS).
TES группу избирательно можно отщепить действием НОАс в водном ТГФ, 2% HF или HF в пиридине.
Введение. Ввести TES группу можно Et3SiCl в присутствии имидазола. Триэтилсилилтрифлат в пиридине или 2,6-лутидине используется для защиты β-ОН группы альдегидов, кетонов, сложных эфиров.
Метиловые эфиры Метокси-группа очень стабильная и требует для своего снятия жестких условий.
Получение метиловых эфиров: Для получения метиловых эфиров используются методы, которые вам уже известны. Это различные варианты реакции Вильямсона, в которой спирт реагирует с MeI, (MeO)2SO2 или CF3SO2Me в присутствие подходящих оснований. Для О-метилирования фенолов используют K2CO3 и (MeO)2SO2, для алифатических спиртов NaH, (Me3Si)2NLi.
В качестве метилирующего средства часто используют диазометан в присутствии протонных кислот HBF4 или кислот Льюиса (ZnCl2, BF3, SnCl4).
Бензиловые эфиры – Bn Как и метиловые эфиры – бензиловые стабильны. Они устойчивы в кислых и щелочных условиях, устойчивы к действию гидридов металлов и мягких окислителей. При повышенных температурах и особенно в присутствии кислот Льюиса бензиловые эфира расщепляются LiAlH4. n-BuLi в ТГФ может отщеплять бензильный протон из бензильной группы. s-BuLi и t-BuLi более подходят для таких целей. Получение: Методы получения аналогичны получению метиловых эфиров. Это алкилирование алкоксидов металлов с бензилбромидом или хлоридом. Алкоксиды обычно генерируют NaH и KH, при это часто необходимо действие катализаторов, KI или Bu4NI добавляют, чтобы из бромидов и хлоридов генерировать in situ иодид. В этих условиях даже третичные спирты могут бензилироваться.
Можно использовать BnBr в присутствии Ag2O.
Естественно можно использовать PhCHN2 при -40°С в присутствии HBF4 CH2Cl2.
Расщепление: Каталитическое гидрирование мягкий метод снятия O-Bn защиты. Катализаторы Pd/C в ТГФ, Ni-Ренея. Естественно при таком методе снятия в молекуле не должно быть кратных связей и групп, которые гидрируются и отравляют катализатор.
Можно осуществлять снятие защиты Na или Li в NH3.
Можно использовать кислоты Льюиса в CH2Cl2 или ClCH2CH2Cl и TMSI (SnCl4, BCl3, FeCl3, BF3).
Тритильные группы – Tr Эта защитная группа используется в синтезе нуклеотидов и нуклеозидов.
Получение: Защищает избирательно первичную OH-группу, вторичные и третичные не реагируют. Используют Ph3CCl в пиридине или диметиламинопиридин.
Можно использовать и TrOTf трифлат.
Расщепление: Использются протонные кислоты HCOOH в эфире. В этих условиях сохраняются изопропенилиденовые группы и tBuMe2Si: 80% кипящей уксусной кислоты, 1 М водной HCl в диоксане.
Для расщепления можно использовать кислоты Льюиса ZnBr2 в MeOH, EtAlCl2-CH2Cl2: FeCl3, а также Na в жидком аммиаке.
Третбутиловые эфиры – t-Bu Стабильны в сильно основной среде и атаке алкиллитиевыми соединениями. Расщепляются в сильно кислой среде по E1еханизму с элиминированием изобутилена. Это направление обусловлено стабильностью третбутилкатионы. Аналогичная трансформация имеет место третбутиловых сложных эфиров и N-третбутоксикарбонил (BOC) производных.
Получают действем избытка изобутилена на спирт в CH2Cl2 в присутствии концентрированной серной кислоты.
Защитные группы для диолов Используются бензилиденацетали, изопропенилиденацеталь, циклогексилидены или циклопенталиденацетали.
Эти защитные группы используются для 1,2- и 1,3-диолов. Связано это с тем, что при этом образуются пяти или шестичленные циклы с 1,3-расположением атомов кислорода.
С другой стороны ацетальную защиту можно рассматривать как защитные группы для карбонильных соединений. Свойства ацеталей вам хорошо известны (химия углеводов).
Зная свойства ацеталей, можно говорить, что эти защитные группы должны быть устойчивы в щелочных условиях и достаточно легко разлагаются в кислых условиях. Бензилиденацетали Устойчивы в большинстве достаточно сильных основаниях, мягких оксидантов и гидридов металлов (в отсутствии кислот Льюиса). Бензилиденацетали легко атакуются N-бромсукцинимидом и озоном.
Бензилиденацетали нестабильны к действию сильных оснований – соединений алкиллития, они подвергаются гидрогенолизу в присутствии Pd или Pt. Кислоты Льюиса также расщепляют бензилиденацетали.
Получение: Имеется два метода синтеза такого рода производных 1,3-диоксанов и 1,3-диоксоланов: 1) реакция диола с бензальдегидом (и его замещенными по кольцу производными) в присутствии протонных кислот и кислот Льюиса (чаще всего ZnCl2); 2) реакция диола с диметилацетилем бензальдегида (ацетальный обмен) в условиях кислого катализа.
Мы можем ожидать для полигидроксипроизводных образование как пятичленного (1,3-диксоланового цикла), так и шестичленного (1,3-доискинового).
В отсутствии заместителей в полиоле мы в праве ожидать образование диоксана, так это термодинамически более стабильный продукт.
Строение защищенного продукта очень сильно зависит от температуры, заместителей и условий проведения, строения карбонильного соединения.
Альдегиды имеют тенденцию давать шестичленные циклы, кетоны (ацетон, циклогексанон) – пятичленные.
В кетонах аксиальный заместитель ацетального центра дестабилизирует соответствующий 1,3-диоксан. В бензалиденацеталях 1,3-диолов Ph занимает экваториальное положении, предсказать геометрию (ориентацию Ph) в случае диоксоланов сложнее.
Снятие бензилиденовой защиты:
1) Каталитическое гидрирование. Мягкие условия. 2) Na и Li в жидком аммиаке в присутствии доноров протонов
3) Обычный тривиальный метод – кислый гидролиз
Обращаю ваше внимание на то, что ацетальную защиту при наличии нескольких групп можно снимать избирательно. Для снятия бензилиденовой защиты используют восстанавливающие реагенты, чаще всего комплексные гидриды, которые позволяют снимать защиту только с одной ОН-группы в то время как вторая остается защищенной в виде бензилового эфира
Введение алкокси-групп в фенильное кольцо резко уменьшает стабильность ацеталей. Так р -метоксибензилиденацетали примерно в 10 раз быстрее гидролизуются, чем бензилиденацетали. Для них можно легче подобрать условия региоселективного расщепления под действием комплексных гидридов.
Изопропенилиденацетали. Это чаще всего используемые защитные группыдля 1,2- и 1,3-диолов. Введение ацетальной защиты: Самый распространенный метод: диол+ацетон+кислота. В тех случаях, когда требуется удалить воду используют молекулярные сита (3-4 Å) или CuSO4 (безводный). Диоксоланы предпочитают диоксанам. Цис-связанные системы (диоксоланы) предпочтительнее.
Можно использовать ацеталь ацетона
Для получения транс-сочлененных ацеталей используют 2-метоксипропен в ДМФА в присутствии п-ТСК
Силильная защита
Защитные группы для азота N-ацильные производные, N-сульфонильные производные, N-сульфенильные производные, N-алкильные производные, N-силильные производные, имино-производные.
N-ацильные производные
Встречается 10 N-ацильных защитных групп: фталимидная, трифенилацетамидная, метокси- и этоксикарбонильная, третбутоксикарбонильная, бензилоксикарбонильная, аллилоксикарбонильная, 9-флуорилметоксикарбонильная, 2-(триметилсилил)этоксикарбонильная, 2,2,2-трихлорэтоксикарбонильная. Прикстически все защитные ацильные группы превращают азот в карбамоильные производные. Все карбаматы легко гидролизуются до карбаминовой кислоты, которая нестабильна и распадаясь дает амин и СО2.
Фталимиды Phth и Pht Стабильны к действию Pb(AcO)2, 30% Н2О2, SOCl2, HBr в AcOH, большинству реагентов, используемых для окисления спиртов в том числе реактиву Джонса, OsO4, большинству методов получения ацеталей. Они устойчивы в условиях перэтерификации ацетатных групп MeONa в MeOH. Лабильны к Na2S·9H2O, гидридам металлов, к действию пиперидина, используемого для снятия 9-флуорилметоксикарбонильной группы (Fmoc).
Введение защитной группы 1) Первичные амины с фталевым ангидридом в CHCl3 при 70°С 4ч. 2) 3) N-этоксикарбонилфталимид
Расщепление. Расщепление как и в методе Габриеля при синтезе первичных аминов. Гидрогенолиз. Его можно использовать для удаления фталимидной защиты в присутствии ВОС-защиты
Можно использовать для снятия защиты MeNHNH2, в этом случае не затрагивается Et3SiO защита.
Трифторацетамидная защита
Получение: (CF3CO)2O в CH2Cl2 в присутствии Et3N или пиридина. Для селективного трифторацетилирования первичной аминогруппы в присутствии вторичной используют N-трифторацетилсукцинимид. Следующий пример показывает возможность трифторацетилирования аминогруппы в присутствии гидроксильной.
Расщепление проводят в щелочной среде: LiOH, K2CO3. N-сульфиниламиды
1) Сульфониламидная группа уменьшает чувствительность пирролов и индолов к электрофильному замещению и окислению. 2) Замещение NH в индолах, пирролах, карбазолах и имидазолах на арилсульфамидную обусловливает литирование гетероцикла.
Введение защитной группы. В индолы, пирролы, имидазолы и т.д. арилсульфонильную группу вводят с помощью сульфонилхлорида в присутствии подходящего основания. Ниже приведены примеры BuLi, гидрид металла в ТГФ и условия межфазного катализа.
В этих условиях сохраняется ВОМ-защита, которая в стандартных условиях PTSA снимается.
Реакция ацетолизации довольно часто в случае циклизации винилкетонов сопровождается миграцией кратной связи.
Иногда эту миграцию используют в синтетических целях.
Снятие защиты: ацетали расщепляются кислотами Льюиса со скоростями, зависящими от природы металла. Большинство цикличесикх ацеталей выдерживают действие MgCl2 и ZnCl2 при низких температурах, но с повышением температуры появляется опасность ращепления.
Сильные кислоты Льюиса Al, Ti, B, R3SiCl вызывают расщепление ацеталей. Например, в присутствии солей Ti и И реактивы Гриньяра, купраты, диалкилцинк, аллилсилаты замещают один из атомов кислорода в ацеталях.
Ацетали ароматических карбонильных соединений подвергаются гидролизу и восстанавлению при растворении металлов.
Ацетали стабильны и устойчивы к действию гидридов металлов и литийорганических соединений, действию водных и спиртовых растворов сильных оснований, каталитическому гидрированию и действию металлов Na или Li в жидком аммиаке.
Основной метод снятия ацетальной защиты это кислый нидролиз. Следует учитывать, что 1,3-диоксановые производные кетонов гидролизуются труднее, чем 1,3-диоксолановые. В тоже время 1,3-диоксолановые производные альдегидов устойчивее 1,3- диоксановых. Очень часто процесс снятия ацетальной защиты происходит в процессе хроматографирования.
Иногда для снятия защиты используют PdCl2(MeCN)2 в ацетоне или FeCl3 на силикагеле.
Защиты карбосильных групп.
Для защиты карбоксильной группы имеется 85 защитных групп (защитные группы в органическом синтезе) и 72 из них эфирные в той или иной форме. Большинство защитных групп вводят с использованием водорода с основными реагентами.
Этерефикация карбоксильной группы кислотой.
Можно выделить следующие традиционные методы этерефикации. 1. Прямое получение из кислот и эфиров. Спирт используется в большом избытке, поэтому он должен быть дешевым и легко кипящим.. Этот метод хорош для метиловых, этиловых,........эфиров, т.к. реакция этерефикации требует применения сильных кислот в качествое катализаторов, то субстрат должен быть стабилен к действию кислот. 2. Реакция хлорангидридов и ангидридов кислот со спиртами. Это вероятно, наиболее широко используемый метод в широком интервале субстратов и условий. Поэтому всегда можно подобрать условия приемлимые для других функциональных групп (не затрагивающие другие группы). Реакцию ведут в присутствии мягких оснований – триэтиламин или пиридин. Стерически трудные спирты реагируют очень слабо. В этом случае модно добавить ДМАР (диметиламинопиридин). И тогда этерефикация значительно ускоряется. 3. Реакция солей кислот с алкилгалогенидами. Этот метод используется для приготовления метиловых, этиловых, аллиловых и бензиловых эфиров, т.к. реакция протекает по SN2 механизму, то она ограничивается первичными галогенидами. Использование солей цезия или тетраалкиламмониевых солей является наиболее эффективным. 4. Реакция карболоновых солей с олефинами. Этот метод применятеся чаще всего для пригтовления третбутиловых эфиров.
Реакцию проводят в присутствии сильной минеральной кислоты. 5. Реакция карбоновых кислот с диазоалканами. Этот метод один из самых мягких и эффективных методов этерефикации. Чаще всего использутеся дя получения метиловых и бензиловых эфиров, т.к. соответствующее диазосоединение получается легко.
Кроме этих традиционных методов рассмотрим новые методы этерефикации. а) использование дициклогексилкарбодиимида (DCC) для активации карбонильной группы. В этом случае реакция идет через О-ацилизомочевину.
По этой реакции (Стеглин) этерефицируются первичные, вторичные и третичные спирты при 20°С. Этот метод можно использовать для этерификации спиртами очень чувствительными к действию оснований.
б) Этерефикация Ямагуши через смещанные ангидриды 2,4,6-трихлорбензойной кислоты. Впервые из кислот и 2,4,6-трихлорбенлилхлорида в присутствии DMAP или триэтиламина образуется смешанный ангидрид, который и этерифициреут спир. Этот метод применим для этерификации вторичных спиртов без рацемизации соседнего хирального центра. Этот метод часто используется для получения макроциклических лактонов, например, 10-членных.
PMB-n-метоксибензил DMB- 3,4-диметоксибензил
в) Этерификация, активированная N-амилгалопириниевыми солями. Можно использовать для синтеза 9-членных циклов.
г) Этерификация Митсунобу. Реакция протекает в нейтральных условиях, используется диэтилазодикарбоксилат. Процесс напрвлен на активацию ОН группы. Этерификация вторичных спиртов протекает с обращением конфигурации. Чаще всего используется в синтезе природных продектов, например, ингибитора глоэспорона.
Метиловые эфиры.
Получение. 1. Реакция с дазометаном. Последний получают реакицей N-метил-N-нитрозо-nтолуолсульфамида с КОН и исползуют в эфирном растворе. Диазометан летуч, токсичен, взрывоопасен. Но несмотря на это, метод используется в случае небольших загрузок. Диазометан в растворе желтый, и при встеплении в реакцию раствор обесцвечивается. В последнее время используют периметилимидазометан. 2. Этерификация кислот в метаноле в присутсвии 2 экв Me3SiCl используется для этрификации аминокислот.
В начале генерируется триметилсилиловый эфир, который in situ превращается в меттиловый эфир.
Na, K, CsCOMe и тетраалкиламмониевык соли карбоновых кислот и метилиодид в ДМФА.
Использование N,N’-диалкил-О-метилизомочевин.
Этот тип реакции позволяет осущетсвить этерификацию кислоты в присутствии незащищенных ОН-групп.
Снятие метильных защитных групп.
Обычный метод – гидролиз, катализируемый гиороксилами металлов или карбонатами в метанле или ТГФ, - ограничивается настабильностью субстратов в основных условиях.
Следующий пример иллюстрирует селективный гидролиз сложноэфирной группы.
Карбоксилат анион является плохой уходящей группой, однако галогенид, тиолят или уианид анионы, чью нуклеофильность модно повысить, исопльзуя диполярные апротонные расворители, атакуют метиловые, этиловые и бензиловые эфиры при обычных температурах. Метиловые эфиры в 70 раз быстрее расщепляются по сравнению с соответствующими этиловыми эфирами, что можно использовать для селективного расщепления.
Понизить температуру расщепления метиловых эфиров можно, используя высокую нуклеофильность тиолят аниона в диполярных растворителях или используя тиоэфиры в присутствии кислот Льюиса.
При этом ОМ
|
||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-30; просмотров: 1458; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.117.153.38 (0.339 с.) |
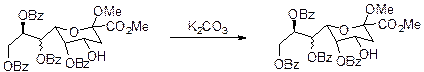
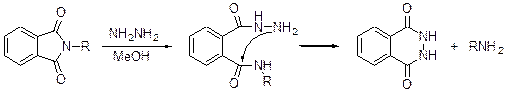


 MeOCH2OMe (MOM) - легко расщепляются в кислых условиях в PhOCH2OMe (BOM) процессе гидролиза.
MeOCH2OMe (MOM) - легко расщепляются в кислых условиях в PhOCH2OMe (BOM) процессе гидролиза.





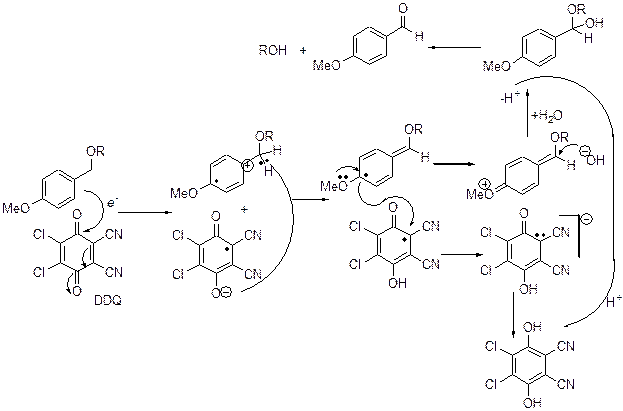


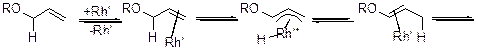
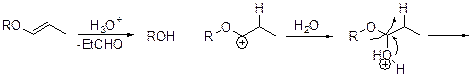








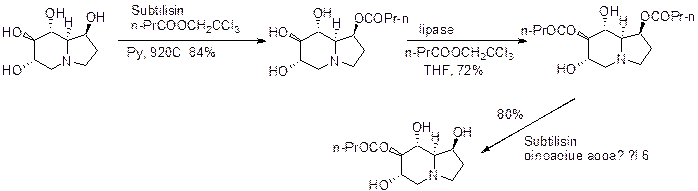



 , гексаметилдисилазан (HMDS)
, гексаметилдисилазан (HMDS)



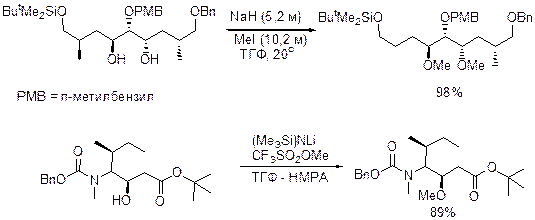

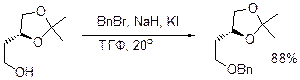
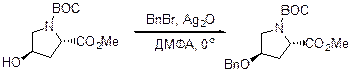
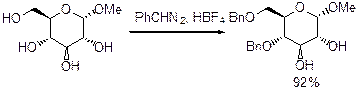
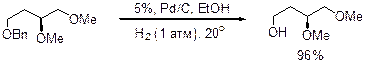
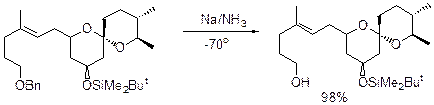

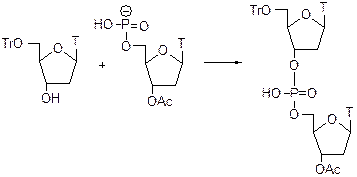

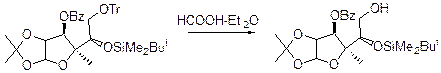
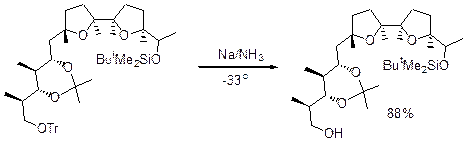
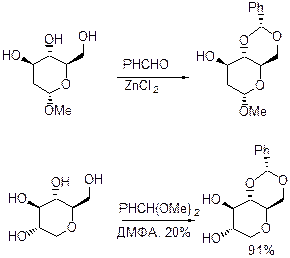









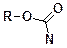
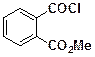 в присутствии оснований
в присутствии оснований