
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Види стійкості дисперсних системСтр 1 из 5Следующая ⇒
Теорія будови ПЕШ Першу найпростішу модель ПЕШ запропонували Гельмгольц та Перрен. Згідно з нею заряди розташовані у вигляді двох рядів різнозарядних іонів на межі зіткнення фаз. Товщина шару вважалася близькою до молекулярних розмірів або розмірів сольватованих іонів. Падіння потенціалу ПЕШ проходить лінійно на відстані δ до нуля (відповідно до теорії плоского конденсатора) (рис. 5.8). де qs – щільність заряду поверхні (заряд одиниці площі поверхні), δ – товщина шару Гельмгольца, ɛ, ɛ0 – діелектрична проникність середовища (ɛ – відносна діелектрична проникність, ɛ0 – електрична стала, 8,854×10-12 [Ф/м]. Модель Гельмгольца-Перрена справедлива лише за відсутності теплового руху йонів. У реальних же умовах є певне співвідношення сил електростатичної взаємодії йонів і їхнього руху. При цьому йони прагнуть рівномірно розподілятися у всьому об’ємі рідкої або газоподібної фази. Виходячи з цього Г. Гуї і Д. Чепмен запропонували свою модель ПЕШ.
Модель ПЕШ Гуї-Чепмена Припускає розпушену (дифузну) будову ПЕШ і те, що всі протийони знаходяться у його дифузній частині. При чому поблизу поверхні, де переважає електричне поле, концентрація протийонів більша, потім зі збільшенням віддалі від поверхні вона поступово зменшується. Згідно з уявленням Гуї та Чепмена, виникнення електрокінетичного потенціалу пояснюється тим, що при відносному переміщенні фаз шар рідини певної товщини міцно утримується на твердій поверхні. Стрибок потенціалу площині розриву такого шару рідини відповідає ξ-потенціалу. Дифузна побудова ПЕШ дала змогу з’ясувати причину електрокінетичних явищ, а також вплив концентрації і зарядів іонів електролітів на величину ξ-потенціалу. Недоліком теорій Гельмгольца-Перенна і Гуї-Чепмена було те, що вони уявляли йони як точкові заряди, що не мають власних розмірів. Нехтування розмірами спричинювало те, що не враховувалась мінімальна товщина шару ПЕШ. Крім того, теорія Гуї-Чепмена не з’ясовувала дії йонів різної природи і ступеня окиснення (валентності), зв’язаної з їхньою специфічною адсорбцією на міжфазовій поверхні, та явище перезарядки колоїдних частинок.
Теорія ПЕШ Штерна Сучасна теорія будови ПЕШ – узагальнена модель Штерна.
Штерн припустив, що ПЕШ складається з двох частин: - внутрішньої – адсорбційний шар; - зовнішньої – дифузний шар Гуї. Внутрішню частину ПЕШ, що знаходиться безпосередньо біля міжфазової поверхні, Штерн уявляв як адсорбційний монойонний шар, товщиною не менше двох радіусів іонів. Стрибок потенціалу на межі адсорбційного і дифузійного шарів Штерн назвав φs-потенціалом, або адсорбційним потенціалом. Введений Штерном потенціал часто називають штернівським. Шар протийонів Штерн поділив на дві частини: одна частина входить до адсорбційного шару, а друга – знаходиться за адсорбційним шаром у дифузній частині ПЕШ. Товщина цього шару (λ) може бути значною і залежить від властивостей і складу системи. Потенціал в адсорбційному шарі зменшується лінійно від φs до φs, як у плоскому конденсаторі. Потенціал у дифузній частині ПЕШ зі збільшенням відстані (х) змінюється експоненціально від φs до 0. Згідно з уявленням Штерна заряд адсорбційного шару складається із заряду йонів, адсорбованих за рахунок електростатичного адсорбційного потенціалу FZφ, як і за рахунок потенціалу специфічної адсорбції. Штерн припускав, що поверхня має певну кількість адсорбційних центрів, кожен з яких взаємодіє з одним протийоном. Оскільки теорія Штерна враховує наявність щільного адсорбційного шару йонів, то це дає можливість виявити вплив їхньої гідратації на поверхневу щільність зарядів цього шару. А врахування специфічної адсорбції йонів дозволяє з’ясувати перезарядку поверхні при наявності у розчині протийонів з більшою адсорбційною здатністю. Краще адсорбуються і ближче підходять до поверхні менш гідратовані йони, які активніше компенсують поверхневий потенціал і відповідно у дифузному шарі їх буде менше. Специфічна адсорбція залежить від спорідненості адсорбованих іонів і поверхні, та їхньої можливості утворювати поверхневі сполуки, що не дисоціюють. Наприклад, на кристалах добре адсорбуються з розчину ті йони, що утворюють з іонами кристала нерозчинні сполуки. Більші адсорбційні потенціали мають багатозарядні йони Cu2+, Ca2+, Fe3-, Th4+, та інші.
Іони, що добре адсорбуються в щільному шарі, іноді здатні не тільки повністю компенсувати поверхневий потенціал, але й створювати надлишковий заряд зі знаком заряду протийонів. Це явище називають перезарядкою. Перезарядка призводить до зміни протийонів у дифузному шарі на йони з зарядом протилежного знаку. Явище перезарядки відбувається завдяки адсорбції на поверхні йонів, однойменних з протийонами. При перезарядці стрибки потенціалу φs і потенціалу адсорбційного шару φs мають різні знаки. Теорія Штерна дала пояснення явищу перезарядки. Згідно з теорією Штерна площина «ковзання» знаходиться за площиною адсорбційного шару. Вчення про ПЕШ і далі розвивається. Воно має значення для з’ясування багатьох практично важливих процесів, приміром: коагуляція йонів, флотація. йонний обмін тощо.
Електрокінетичні явища …полягають у взаємозв’язку між електричними і кінетичними властивостями дисперсних систем. ПЕШ на межі розділу дисперсної фази і дисперсійного середовища зумовлює можливі взаємодії дисперсних систем з електричним полем. Під дією електричного поля можливі відносні переміщення двох фах. З іншого боку, при переміщені цих двох фаз одна відносно іншої, може виникати різниця потенціалів. Ці ефекти називають електрокінетичними явищами. Усі електрокінетичні явища пов’язані з існуванням на межі розділу фаз ПЕШ.
Електрокінетичний потенціал Межа, відносно якої дисперсійна фаза і дисперсійне середовище зсуваються одне відносно іншого, знаходиться або у дифузійному шарі протийонів, або на відстані δ від поверхні. Хоча φs є чітко визначена величина (це штернівський потенціал – стрибок потенціалу на межі адсорбційного і дифузійного шарів), виміряти його практично неможливо. Експериментально можна визначити іншу, близьку до φs величину – електрокінетичний потенціал, або дзета-потенціал ξ. Цю величину можна визначити як стрибок потенціалу, що виникає в площині «ковзання» при відносному переміщенні фаз. У загальному випадку ξ-потенціал менший за φs-потенціал. Ця різниця тим більша, чим менша дифузійна частина ПЕШ, та чим менша величина λ (товщина диференційного шару). Отже, усі чинники, що впливають на λ, змінюють значення ξ-потенціалу. Наприклад, зменшення товщини дифузійного шару, а отже і ξ-потенціалу відбуваються при: а) зниження температури; б) введенні в систему індиферентного електроліту, який специфічно не взаємодіє з поверхнею; в) а також, збільшенні заряду його йонів; г) зменшенні діелектричної сталої середовища, наприклад, при додаванні до гідрозолів спиртів, егерів, естерів тощо. ξ-потенціал можна виміряти, вивчаючи електрокінетичні явища, а саме: - електроосмос; - електрофорез; - потенціал перебігу; - потенціал седиментації.
Електроосмос При дослідження електролізу води було помічено підняття рівня водив у посудині з негативно зарядженим електродом, якщо в U-подібній трубці нижня частина засипана кварцовим піском. Підняття рідини в циліндрі з негативним електродом відбувалося доти, доки не встановлювалася певна різниця рівнів – рівновага з гідростатичним тиском. Подібний дослід, але без заповнення низу трубки піском, не давав такого ефекту. Отже, можна було зробити висновок (Ф. Рейсс): при контакті з частинками кварцового піску рідина заряджається.
З часом явище спрямованого переміщення дисперсійного середовища відносно нерухомої дисперсної фази в постійному електричному полі одержало назву електроосмосу. Спрямоване переміщення рідини вказує на знак ξ-потенціалу, т.і. на знак заряду твердої поверхні на межі з рідиною. Вимірюючи швидкість течії рідини, можна розрахувати стрибок потенціалу на поверхні «ковзання», за рівнянням Гельмгольца-Смолуховського, які вивели його на підставі уявлень про ПЕШ як плоский конденсатор: де η – в’язкість рідини; – лінійна швидкість течії рідини; H – напруга зовнішнього електричного поля (градієнт потенціалу); E – зовнішня різниця потенціалів; l – віддаль між електродами; ɛ – діелектрична проникність рідини; ɛ0 – електрична стала, 8,854×10-12 [Ф/м]. Швидкість руху дисперсійного середовища, віднесену до одиниці напруги електричного поля, називають електроосматичною швидкістю, звідси: лінійну швидкість течії рідини доцільно замінити на об’ємну швидкість течії рідини. Користуючись законом Ома, перетворюють член, де V – об’ємна швидкість рідини; – питома електропровідність; I – сила струму. Отже, підставивши H0/H в рівняння для ξ одержуємо розрахункову формулу: В це рівняння входять величини, які можна виміряти експериментально. Крім вимірювання ξ-потенціалу, метод електроосмосу має велике практичне застосування в процесах дисводнення пористих матеріалів і концентруванні колоїдних систем.
Електрофорез Явище спрямованого переміщення частинок дисперсної фази в електричному полі. На початку ХІХ ст. Ф. Рейсс, вивчаючи електроліз води, провів такий експеримент. Він заповнив водою скляні трубочки, занурені перед цим у глину. Після накладання електричного поля він спостерігав переміщення частинок глини в рідині до позитивного електрода. Електрофорез можна спостерігати лише в седиментаційних стійких дисперсійних системах. Рухливість частинок в електричному полі зумовлена тим, що при накладанні зовнішнього електричного поля, відбувається розрив ПЕШ по площині «ковзання». Внаслідок цього частинка одержує певний заряд і переміщується до електрода з зарядом, протилежним заряду цієї частинки. При цьому протийони дифузійного шару переміщуються до протилежно зарядженого електрода. Швидкість руху частинок у дифузійного шару пропорційна величині ξ-потенціалу.
На відміну від електроосмосу при електрофорезі можна безпосередньо вимірювати швидкість руху частинок. Рівняння Гельмгольца-Смолуховського справедливе і для електрофорезу, т.і. Однак, лінійна швидкість руху частинок U0 не може слугувати їхньою характеристикою, бо вона змінюється з напругою зовнішнього електричного поля H. Тому ввели поняття електрофоретичної рухливості Uеф,, яка дорівнює швидкості при одиничному градієнті потенціалу (H = 1).
ξ = Якщо визначити, то
Лінійна швидкість U0 – це фактично відношення лінійного зміщення межі ЗОАЮ S до часу експерименту (t)
Е – зовнішня різниця потенціалів (напруга); l – відстань між електродами. Отже,
т.і. усі величини можна виміряти. Uеф вимірюється в м2/(с×В), а ξ-потенціал частинок колоїдних розчинів знаходиться в межах 1,5-1,75 мВ.
Потенціал перебігу Явище, обернене електроосмосу, є потенціал течії або перебігу. Розділ іонів різних знаків та перенесення зарядів по капіляру призводить до появи на кінцях капіляру різниці потенціалів – потенціалу перебігу. Потенціал перебігу є зокрема джерелом виникнення біопотенціалів. При протіканні крові по капілярах кровоносної системи виникає потенціал перебігу, який відбивається певним чином на електрокардіограмі пацієнта. Можна гадати, що явища виникнення потенціалу перебігу можуть проявлятися при струменевому «друці», т.і. струменевих принтерах. Можливо також в різографах. Потенціал перебігу (U) пропорційний перепаду тиску (p). Рівняння Гельмгольца-Смолуховського для розрахунків ξ-потенціалу за потенціалом перебігу має вигляд: ξ = де η – в’язкість; χ – питома електропровідність; U – потенціал перебігу; p – тиск; ɛ – діалектрична проникність; ɛ0 – електрична стала – 8,854×10-12 [Ф/м]. U визначають як різницю Uі−U0, де U0 – різниця потенціалів між електродами без тиску, Uі – різниця потенціалів при різних значеннях тиску. Визначення ξ-потенціалу цим методом дає достовірніші дані, оскільки в експерименті нема потреби накладати зовнішню різницю потенціалів, яка викликає побічні явища (поляризація, нагрівання).
Потенціал седиментації При осіданні частинок дисперсної фази по висоті посудини виникає різниця потенціалів – потенціал седиментації. Причиною цього явища, оберненого електрофорезу, є також, ПЕШ, який деформується при терті осідаючих частинок об дисперсне середовище. За величиною потенціалу седиментації можна також розрахувати ξ-потенціал. Потенціал седиментації виникає при роботі центрифуги. Можна гадати, що й при ультрафільтрації. Явища виникнення потенціалів перебігу та седиментації спостерігаються при осіданні суспензій та емульсій, при розділенні фаз. На кінцях трубопроводів і апаратів виникають високі різниці потенціалі, які є причиною іскрових розрядів, що спричиняють пожежі та вибухи. Виникнення електростатичного заряду при проході стрічки або через систему валів (друкарство, друкарський офсет) можна також пояснювати певним внеском електрокінетичних явищ в дисперсних систем.
Стійкість і коагуляція дисперсних систем Види стійкості дисперсних систем Фактори стійкості дисперсних систем Теорії стійкості і коагуляції гідрофобних золів Колоїдний захист Коагуляція гідрофобних золів Коагуляція під дією електролітів Правило Шульца-Гарді Кінетика коагуляції Гетерокоагуляція. Взаємна коагуляція Явище привикання золів
Швидкість коагуляції Кінетика швидкості коагуляції розроблена М. Смолуховським (1916), а повільної – Фунеом. Відношення констант швидкостей швидкої та повільної коагуляцій може слугувати мірою стабільності дисперсної системи: Величина W – коефіцієнт стабільності дисперсної системи.
II Початок термоденаміки. Швидко чи повільно (це рівноважна термоденаміка не розглядає), але всяка система направлена до стану істинної рівноваги. Це може служити одним із формулювань другого початку термоденаміки. Його формулюванням можна прийняти і неможливість самовільного переходу теплоти від менш нагрітого тіла до більш нагрітого. Також, згідно другого закону, якщо періодично діюча машина збирає від нагрівача який має температуру ТН, теплоту Q і перетворює її на роботу А, то завжди А ˂ Q і різниця Q-А переходить в вигляді теплоти в холодильнику з температурою Тх (вигляді компенсації за роботу). Робота може досягнути максимального значення: Аmax, a(Q-Amax) – мынымального при зворотному ведены процессу і при використанні ідеальної машини (без тертя); тоді відношення В реальних процесах,які майже ніколи не бувають оборотними, відношення Якщо в системі проходить реакція при постійному тиску і температурі, то зміни ентальпії також може бути запропоновано в вигляді двох доданків: зміни вільної енергії ∆G і зміни зв’язаної енергії ∆L. Першу частину енергії принципово можна перетворити на корисну роботу Аmax при оборотному ізобарно – ізотермічному веденні процесу, а другу – ні. Можна записати.
Де ∆G є зміна вільної енергії Гіббса при постійному тиску і температурі, її називають змінами ізобарно – ізотермічного потенціалу. Зв’язана енергія ∆L виражається добутком абсолютної температури Т на зміни функції стану, яка називається ентальпієюS. ∆L = T∆S. Тоді ∆G = Ізометричні процеси здійснюються при постійному Т, ізобаричний – при постійному тиску (Р), ізохорний - при постійному об’ємі (V). Процеси, в яких може здійснюватися робота, але система не обмінюється теплотою з навколишнім середовищем, називається адіабатичними. Перед тим, як узагальнювати відомості про початки (начала) термодинаміки нагадаємо: Коефіцієнт корисної дії ККД (рос.«КПД»):
де
Власне, це є математичним виразом ІІ початку термодинаміки, яке можна формулювати так: «Тепло не може довільно переходити від холодного (менш теплого) до нагрітого (теплішого) тіла» (визначення Р.Клазузіуса). Серед інших визначень ІІ початку термодинаміки є таке: «Неможливо створити машину, яка б перетворювала усе тепло у роботу», а також «Неможливо створити вічний двигун». Суть ІІ початку термодинаміки випливає з того, що малоймовірно щоб хаотичний тепловий рух молекул перейшов у напрямлений рух. Навпаки, напрямлений рух молекул може повнісю перейти у хаотичний (робота може повністю перейти в теплоту). Хаотичний рух молекул є природнім. Це є причиною того, що різні види енергії прагнуть у теплоту, а теплота передається менш нагрітим тілам. 3.3.3. Ентропія. Отже довільні процеси відбуваються з розсіюванням теплової енергії. Для кількісної характеристики цього процесу Рудольф Клаузіус (1865р.) увів термодинамічну функцію S і назвав її енергією:
Знак нерівності – для необоротних процесів а = - для оборотних. Одиниця виміру. Ентропія – є мірою розсіяної (знеціненої) енергії. Чим більше енергія, тим менша частина енергії може перетворитися в роботу. Отєе, енергія – міра необоротності процесу. Довільно відбувається процеси, в яких ентропія зростає. Це також може бути формуванням ІІ початку термодинаміки dS>0 δQ = TdS Отже TdS ≥ dU + pdv
Це є об’єднаний вираз ІІ та І початків термодинаміки.
Австрійський фізик Людвіг Больцман показав, що ІІ початок термодинаміки має статистичний характер і свідчить лише про ймовірність процесу. Больцман встановив зв'язок між ентальпією S та ймовірністю стану системі W (від німецького W ahrscheinlichkeit.) В англомовній заміст W використовують P від англійського P robability). S = k · ln W k – стала Больцмана (k = Чим більшою кількістю мікрочастинок представлена система, тим більше варіантів розподілу цих частинок, тим вище значення ентропії і тим більш знаціненим буде вмщений у системі запас енергії. Отже, ентропія характеризує відносну цінність енергії, яку має система, тобто характеризує ту частину енергії, яка не перетворюється в роботу.
ІІІ Початок термодинаміки. ІІІ Початок термодинаміки. S T=0 дорівнює 0; lim ST→0 = 0 Дійсно, при T=0 W=1 S = k · lnl = 0
Термодинамічні потенціали Ентропія є функцією, що визначає можливість перебігу довільного процесу в ізольованій системі. Для закритих систем функціями, які визначають таку можливість є термоденомічні потенціали: Ізохорично – ізотермічний потенціал, або вільна енергія Гельмгольца (А) та ізобарично – ізотермічним потенціал, або енергія Гіббса. У більшості випалків (в хімії та поліграфічних технологіях) енергія Гіббса частіше застосовується, оскільки процеси проходять при сталому тиску, а не при сталому обємі.
A=U-TS (∆A=∆U - T∆S) G=H- TS (∆G=∆H - T∆S)
Абсолютні значення Т/д потенціалів. Не відомі. Звичайно користуються величини ∆A Та ∆G, КД (моль). Зв'язок А та G: G = U + pV – TS = A + pV -∆G=-∆A-pV
Зменшення енергії гіббса більше або дорівнює максимальній корисній роботі процесу. G тa А називають також вільною енергією. Т/д потенціали не змінюються в оборотних процесах і зменшуються – в необоротних.
Хімічний потенціал У багатокомпонентних відкритих системах склад і маса кожного компоненту змінюються, що впливає на енергетичний стан кожного учасника і всієї системи загалок. Тому Т/д – потенціали (А та G) для відкритих систем залежить не тільки від зовнішніх умов, але й від кількості кожного з учасників: А= f (T,V,n1,n2,……ni) де n1,n2,…..,ni. кількість молей.. G== f (T,p,n1,n2,……ni) компонентів…? Нагадаймо G=H-TS. (як і: А = U- TS) Звідси: dA = dU – TdS - SdT Оскільки TdS ≥ dU + pdV (об’єднаний вираз І та ІІ начал термодинаміки) dU – TdS ≤ - pdV dA ≤ - pdV - SdT
Неважко показати dG ≤ Vdp - SdT Якщо диференціювати ці фунції за кількістю молей усіх учасників отримуємо
Часткова похідна Г або А усієї усієї системи при зміні вмісту ііншого учасника на 1 моль за умови сталої концентрації усіх інших учасників і зовнішніх параметрів називається парціальним молярним потенціалом ( Отже:
Можна дійти висновку, що Т/д потенціали (вільна енергія характеризують енергетичний стан речовини в багатокомпонентній системі. Для індивідуальної речовини поняття молярного Т/д і хімічного потенціалів є ідентичні, т.т.
Пригадаймо: dG ≤ Vdp - SdT При T=const dG ≤ Vdp Для ідеального газу:
Тому і-го компоненту ідеального газу.
Інтегрування при Т= const Призводить до: μ=RTln
За стандартних умов Р= 1атм = 1.033·105 Па const = μi = Р* - тиск індивідуального газу,
Для реального газу: μi =
f =γp Фугітивність має розмірність тиску і пропорційне йому. Коефіцієнт пропорційності має назву коефіцієнту фугітивності. Для розчинів замість ффугітивності застосовують величину активності а a=γ1·C γ- коефіцієнт активності. Хімічний потенціал реального розчину має вираз μi =
Коефіцієнт активності дозволяє враховувати відхилення властивостей реального розчину від властивостей цього розчину в ідеальному стані. Зміна вільної енергії Гіббса при утворенні ідеального розчину.
∆G = nRT (XAlnXA+XBlnXB) XA, XB - мольні частинки, менші за 1 Тому lnXA, lnXB, ∆G - від’ємні величини. 3.7. Теплота випаровування рідини. Зв'язок між при хованою молярною теплотою випаровування та температурою. Леткість. Температура кипіння розчинників. Розчинення речовин відбувається з виділенням або поглинанням тепла, а іноді із зміною об’єму. Ці явища свідчать про хімічну взаємодію між розчиненою речовиною і розчинником. Експериментально встановлено, що у водному розчині відбувається утворення гідратів (для неводних розчинів – сольватів), які є порівняно нестійкими сполуками розчинених частинок з розчинником. Тепловий ефект, що супроводжує процес розчинення, відносять до одного молю розчиненої речовини і називають молярною теплотою розчинення, відповідно можно говорити про молекулярну теплоту випаровування.
Щоб дана рідина закипіла, тиск її парів повинен бути однаковим із зовнішнім тиском. При барометричному тиску 760мм.рт.ст. (101,3 кПа) для води це буде температура 1000 С. Здебільшого з розчину під час кипіння видаляється тільки розчинник, внаслідок чого концентрація розчину підвищення температури кипіння. Отже розчин кипить не при певній температурі, а в деякому температурою кипіння. Різниця між температурою кипіння розчину і чистого розчиника називається підвищенням температури кипіння розчину (∆t кип). ∆t кип = Де Всяка рідина починає кипіти при тій температурі, при яких тиск її насиченої пари досягає величини зовнішнього тиску. Наприклад, вода під тиском 101,3 кПа кипить при 1000 С тому, що при цій температурі тиск водяної пари якраз дорівнює 101,3 кПа. Якщо ж розчинити у воді яку-небудь нелетку речовину до 101,3 кПа треба нагріти його вище 1000 С. Звідси виходить, що температура кипіння розчину завжди вища за температуру кипіння чистого розчинника.
Вивчаючи кипіння розчинів, Рауль установив, що для розбавлених розчинів неелектролітів підвищення температури кипіння: ∆t кип = Em m – мольно – масова концентрація (молярність); E- ебуліоскопічна сталь, що залежить тільки від природи розчинника, але не залежить від природи розчиненої речовини. Для води ебуліоскопічна стала E дорівнює 0,52,для бензолу E = 2,6. Леткість – це властивість розчину або рідини до випаровування, або перетворення в газоподібний стан.
Адсорбція Довільне зменьшення Адсорбція залежить від природи розчинника і розчинної, а сааме: від значення величини
Рівння адсорбції Гіббса Адсорбція на межі поділу рідина-газ, рідина-рідина. Як з’ясував Гіббс, розподіл розчиненої у розчині речовини відбувається так,що при цьому досягається максимальне зменшення поверхневого натягу.
Рівняння Гіббса є матиматичним обгрунтіванням загального правила: речовина, яка зменшує поверхневий натяг, концентрується у поверхневому шарі, і навпаки. Якщо σ зменьшеться при збільшенні с, то Граничне значення
Корисними на практиці є правило Дюкло-Таубе: Поверхнева активність (g) дифільних молекул (жирнрих кислот, спиртів т.і.) У водних розчинах однакової концентрації збільшується у 3-3.5 рази зі збільшенняч довжини карбогідроненого радикалу на одну (-C
Користуючись правилом Дюкло-Таубе можна підібрати ПАР потрібної поверхневої активності.
Для визначення адсорбції будують ізотерму поверхневого натягу Г. (рис.4.6, 4.7)
Із залежностей (рис.4.6) знаючи кут нахулу (АЛЬФА), визначають адсорбцію:
При високих концентраціях Г досягає свого граничного значення (Г∞) і далі не змінюється (рис. 4.7) (Г ∞) - постійна для всіх членів гомологічного ряду ПАР.
4.3.5. Адсорбція на межі тверде тіло-газ. Усі випадки поглинання газів і пари твердими тілами називають сорбцією. Якщо процес сорбції відбувається тільки на поверхні, то його називають адсорбцією. Адсорбцією – явище, коли речовинна, що поглинається поверхнею, дифундує у середину (об’єм) поглинаючої речовини і розподіляється по об’єму. Верде тіло, на поверхні якого відбувається адсорбція, називають адсорбентом, а речовину, яка адсорбується адсорботивом або адсорбатом. Процес, протилежний адсорбції – десорбція. Десорбція – це відокремлення адсорбованих молекул з поверхні шару адсорбенту. Коли швидкість десорбції зрівнюється зі швидкістю адсорбції наступає адсорбційна рівновага ( Отже розрізняють 2 види адсорбції: хімічну та фізичну. Першу називають також хемосорбцією. Фізична адсорбція зумовлена взаємодією поверхневих молекул адсорбенту з молекулами адсорбату. При цьому молекули адсорбату не втрачають своєї індивідуальності. Адсорбція відбувається тільки на певних ділянках адсорбенту, які мають надлишкову поверхневу енергію порівняно з площею поверхні. Для фізичної адсорбції характерна швидка оборотність процесу адсорбція-десорбція, зменшення адсорбції з підвищенням температури. Хімічна ж адсорбція часто зростає. Адсорбція залежить від тиску газу над поверхнею твердого адсорбенту. 4.3.5.1 Рівняння адсорбції Ленгмюра. Адсорбція газів твердим адсорбентом (позначають також А) описується рівнянням Ленгмюра:
Р- тиск газу (пари). Ізотерма адсорбції Ленгмюра може нагадувати ізотерму адсорбції Гіббса (рис.4.7, 4.8) При низьких Р рівняннях Ленгмюра перетворюється у вираз: Тобто поверхнева концентрація газу Г прямо пропорційна тискові Р (рис.4.7, ділянка (І) При високих Р Р>>К і Г= 4.4 Адсорбція на межі поділу тверде тіло-розчин. Ізотерми адсорбції розчинених речовин із розчинів аналогічні ізотермам адсорбції газів. Для розведеннх розчинів ці ізотерми досить добре підпорядковуються рівнянню Ленгмюра. Адсорбція з розчинів порівняно з газовою адсорбцією має певні особливості: 1) На поверхні шару разом з молекулами розчиненої речовини можуть адсорбуватись і молекули розчинника. Тому можливі два різновиди адсорбції: додатна і від’ємна. В останньому випадку молекули розчинника краще адсорбуються за молекули розчиненої речовини. 2) Адсорбція з розчинів відбувається повільніше від адсорбції газів. Для прискорення встановлення адсорбційної рівноваги систему перемішують. 3) Підвищення температури розчину зменшує адсорбцію, але не так сильно, як адсорбцію газів. В залежності від природи розчиненої речовини (молекули чи йони) адсорбцію поділяють на молекулярну та йонну. Таким чином, на адсорбцію з розчинів впливають природа розчиненої речовини, природа розчинника, природа адсорбенту. 4.4.1 Адсорбція електролітів. Електроліти у водних розчинах знаходяться у вигляді йонів. Тому адсорбцією сильних електролітів з розчинів називають йонною. Йонна адсорбція – це хімічна реакція між йонами розчиненої речовини і твердою поверхнею адсорбенту. Енергії нового хімічного зв’язку недостатньо для того, щоб відірвати поверхнево активні атоми адсорбенту. Тому зберігаються зв’язок нової сполуки з твердим адсорбентом. Адсорбція йонів відбувається за двома основними механізмами: 1 – вибіркова адсорбція йонів на кристалах. 2 – еквівалентна або іонообмінна адсорбція. 4.4.2 Вибіркова адсорбція. Вибіркова адсорбція визначається вибірковістю адсорбції катіона або аніона. Правила вибіркова адсорбції сформульовано Панетом і Феянсом. Перше правило: Кристалічну гратку адсорбенту добувають ті йони, що входять до її складу, ізоморфні з її йонами й утворюють з йонами цієї гратки важкорозчинні сполуки. Друге правило: На твердій поверхні адсорбенту адсорбуються тільки ті йони, знак заряду яких протилежний заряду поверхні адсорбенту. 4.5. Змочування. Поверхневий натяг і міжмолекулярні взаємодії всередині фаз змочують процеси змочування і розтікання краплі рідини на тверді або рідинній поверхні, а також явища когезії та адгезії. Змочування. Маленька крапля рідини на твердій поверхні може набути різної форми: або близьку до сферичної (як крапля води на поверхні парафіну) вбо кляксу, оскільки розтікається по поверхні твердого матеріалу, подібного до краплі води на поверхні чистого скла. В першому випадку тверда поверхня не змочується рідиною, а в другому випадку змочується. Явище змочування впливає на перебіг багатьох процесів в природі та техніці, зокрема в поліграфічних технологіях. Це явище є початковоюб стадією взаємодії рідин з твердими тілами. За кількістю фаз, які беруть участь у процесі, можна виділити два різновиди змочування: Імерсійне та контактне.
|
|||||||||
|
|
Последнее изменение этой страницы: 2016-09-19; просмотров: 645; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.161.116 (0.199 с.) |
 , т.і.
, т.і.  звідси:
звідси:


 , яка має нахву коефіцієнтом корисної дії, такоє досягае максимальне значення, яке дорівнює:
, яка має нахву коефіцієнтом корисної дії, такоє досягае максимальне значення, яке дорівнює: 
 завжди менше відношенню
завжди менше відношенню  . Це означає, що в найкращому випадку частину забраної від нагрівача енергії в вигляді теплоти може бути використана як корисна частина в вигляді роботи Аmax. Ця величина характеризує так звану вільну енергію, а інша частина ні за яких умов не може бути перетворена на роботу – ця зв’язана енергія.
. Це означає, що в найкращому випадку частину забраної від нагрівача енергії в вигляді теплоти може бути використана як корисна частина в вигляді роботи Аmax. Ця величина характеризує так звану вільну енергію, а інша частина ні за яких умов не може бути перетворена на роботу – ця зв’язана енергія.
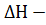 T∆S і G = Н - TS.
T∆S і G = Н - TS.
 ,
, - температура нагрівника.
- температура нагрівника.  - температура холодильника.
- температура холодильника.
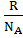 )
)
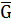 i,
i,  i) даного іншого учасника, або хімічним потенціалом.
i) даного іншого учасника, або хімічним потенціалом.

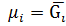 i
i 
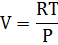
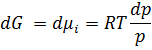
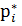 + const
+ const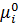


 футивність (замічть тиску). Фугітивніть – це величина тиску реального газу, при підстановці якої у рівняння можна застосувати для описання поведінки реальних газів.
футивність (замічть тиску). Фугітивніть – це величина тиску реального газу, при підстановці якої у рівняння можна застосувати для описання поведінки реальних газів.
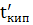 - t кип
- t кип  відбувається при абсорбції. Адсорбція – довільна зміна концентрації компонентів у поверхневому шарі
відбувається при абсорбції. Адсорбція – довільна зміна концентрації компонентів у поверхневому шарі  у стані рівноваги порівнянно з концетрацією в об’ємній фазі
у стані рівноваги порівнянно з концетрацією в об’ємній фазі  на межі поділу фаз розчин-газ;розчин-розчин. Газ (розчин –тверде тіло). Можна сказати. що це збільшення
на межі поділу фаз розчин-газ;розчин-розчин. Газ (розчин –тверде тіло). Можна сказати. що це збільшення 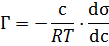
 і адсорбція (Г > 0) буде мати додатнє значення. Така адсорбція позитивна. Якщо ж
і адсорбція (Г > 0) буде мати додатнє значення. Така адсорбція позитивна. Якщо ж 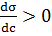 і σ збільшується зі зростанням концентрації розчиненної речовини, то адсорбція буде негативною.
і σ збільшується зі зростанням концентрації розчиненної речовини, то адсорбція буде негативною. при c
при c  0 -(
0 -( g- поверхова активність,
g- поверхова активність,
 -) группу:
-) группу: та
та 

 (4-5)
(4-5) Природа адсорбційних сил може бути: специфічна (хімічна) та неспецифічна (фізична).
Природа адсорбційних сил може бути: специфічна (хімічна) та неспецифічна (фізична).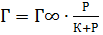 , де: К- коефіцієнт пропорційності,
, де: К- коефіцієнт пропорційності,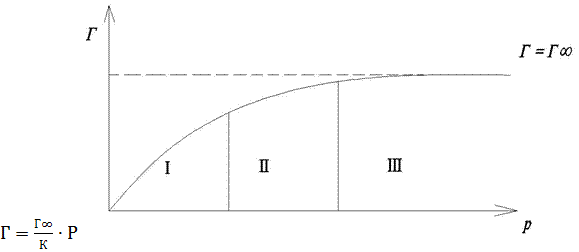
 т.ч. кількість адсорбованих молекул дорівнює кількості активних центрів і адсорбція досягає граничного значення
т.ч. кількість адсорбованих молекул дорівнює кількості активних центрів і адсорбція досягає граничного значення  (рис.4.8. ділянка (ІІІ)
(рис.4.8. ділянка (ІІІ)



