Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Стресс-реализующие системы - симпато-адреналовая система и гипоталамо-гипофизарно-надпочечниковая система.
Воздействие стрессора на организм вызывает формирование очага возбуждения в коре больших полушарий головного мозга, импульсы из которого направляются в вегетативные (симпатические) центры гипоталамуса, а оттуда – в симпатические центры спинного мозга. Аксоны нейронов этих центров идут в составе симпатических волокон к клеткам мозгового вещества надпочечников, формируя на их поверхности холинэргические синапсы. Выход ацетилхолина в синаптическую щель и взаимодействие его с Н-холинорецепторами клеток мозгового вещества надпочечников стимулирует выброс ими адреналина. Курение вызывает повышение концентрации никотина в крови, никотин стимулирует Н-холинорецепторы клеток мозгового вещества надпочечников, что сопровождается выбросом адреналина. Эффекты катехоламинов · Усиление сердечной деятельности, опосредованнное возвуждением b-адренорецепторов сердца. · Расширение сосудов сердца и мозга, опосредованнное возвуждением b-адренорецепторов. · Выброс эритроцитов из депо – обусловлен сокращением капсулы селезенки, содержащей a-адренорецепторы. · Лейкоцитоз – «встряхивание» маргинальных лейкоцитов. · Сужение сосудов внутренних органов, опосредованнное возвуждением a-адренорецепторов. · Расширение бронхов, опосредованнное возвуждением b-адренорецепторов бронхов. · Угнетение перистальтики ЖКТ. · Расширение зрачка. · Уменьшение потоотделения.
· Катаболический эффект адреналина обусловлен активацией аденилатциклазы с образованием цАМФ, который активирует протеинкиназы. Активная форма одной из протеинкиназ способствует фосфорилированию (активации) триглицеридлипазы и расщеплению жиров. Образование активной формы другой протеинкиназы необходимо для активации киназы фосфорилазы b, которая катализирует превращение неактивной фосфорилазы b в активную фосфорилазу а. В присутствии последнего фермента происходит распад гликогена. Кроме этого при участии цАМФ активируется протеинкиназа, необходимая для фосфорилирования гликогенсинтетазы, то есть перевода ее в малоактивную или неактивную форму (торможение синтеза гликогена). Таким образом, адреналин через активацию аденилатциклазы способствует распаду жиров, гликогена и торможению синтеза гликогена.
Активация гипоталамо-гипофизарно-надпочечниковой системы Возбуждение участка коры головного мозга под действием стрессора вызывает стимуляцию гипофизотропной зоны медиальной зоны гипоталамуса (эндокринные центры) и высвобождение гипоталамических рилизинг-факторов, которые оказывают стимулирующее действие на аденогипофиз. Результатом этого является образование и выделение тропных гормонов гипофиза, одним из которых является адренокортикотропный гормон (АКТГ). Органом-мишенью этого гормона является корковое вещество надпочечников, в пучковой зоне которого вырабатываются глюкокортикоиды, а в сетчатой зоне – андрогены. Андрогены вызывают стимуляцию синтеза белка; увеличение полового члена и яичек; ответственны за половое поведение и агрессивность. Другим тропным гормоном гипофиза является соматотропный гормон (СТГ)к эффектам которого относятся: · стимуляция синтеза и секреции инсулиноподобного фактора роста в печени и др. органах и тканях, · стимуляция липолиза в жировой ткани, · стимуляция продукции глюкозы в печени. Третьим тропным гормоном гипофиза является тиреотропный гормон (ТТГ), который стимулирует синтез тиреоидных гормонов в щитовидной железе. Тиреоидные гормоны ответственны за стимуляцию синтеза белка во всех клетках тела, повышение активности ферментов, участвующих в расщеплении углеводов, разобщении окисления и фосфорилирования (увеличения теплопродукции)
Эффекты глюкокортикоидов · Индукция синтеза ферментов – глюкокортикоиды (ГК) проникают через мембрану в цитоплазму клеток, где связываются в комплекс с рецептором (R). Комплекс ГК-R проникает в ядро, где увеличивает синтез РНК-полимеразы, что приводит к ускорению транскрипции мРНК, способствуя образованию белков-ферментов глюконеогенеза. · Мобилизация белковых ресурсов клетки - глюкокортикоиды освобождают свободные аминокислоты из мышечной, лимфоидной и соединительной ткани и почек. · Пермиссивное (разрешающее) действие - особенно четко проявляется в отношении катехоламинов. Катаболический эффект адреналина обусловлен активацией аденилатциклазы с образованием цАМФ, который затем активирует протеинкиназы. Распад цАМФ вызывает фосфодиэстераза, которую ингибируют глюкокортикоиды, тем самым, усиливая эффекты катехоламинов. Кроме того, глюкокортикоиды блокируют ферменты: моноаминоксидазу (МАО), содержащуюся в адренергических окончаниях, и катехол-О-метилтрансферазу (КОМТ), локализующуюся в цитоплазме эффекторных клеток. Эти ферменты вызывают инактивацию катехоламинов. · Увеличение концентрации глюкозы в крови - усиление глюконеогенеза + торможение синтеза белка + пермиссивное действие глюкокортикоидов на эффект (катаболический) адреналина + снижение проницаемости клеточных мембран для глюкозы. · Мобилизация энергетического ресурса клеток - катаболическое действие + активация синтеза ферментов глюкогенеза + торможение синтеза белка, пермиссивное действие глюкокортикоидов на эффект (катаболический) адреналина. · Тормозится воспалительние - глюкокортикоиды стабилизируют мембраны лизосом и блокируют синтез фосфолипаз, препятствуя тем самым выбросу протеолитических ферментов (альтерации) + нормализация повышенной проницаемости сосудов, что препятствует экссудации и выделению медиаторов воспаления + глюкокортикоиды угнетают фагоцитоз. · Снижение иммунитета - торможение синтеза антител (распад белков, репрессия транскрипции) + угнетение фагоцитоза. 9. – Врожденные и наследственные болезни. Мультифакториальные болезни. Генокопии и фенокопии. Классификация врожденных заболеваний в зависимости от срока возникновения. Методы изучения наследственных болезней.
Врожденные заболевания – заболевания, возникающие внутриутробно (пренатально), в период родов (интернатально) и существующие к моменту рождения. Врожденные заболевания могут быть наследственными и ненаследственными, причем более распространены ненаследственные врожденные заболевания. Наследственные заболевания – обязательно сопровождаются поражением генетического аппарата, передаются по наследству. Большая часть наследственных заболеваний проявляется сразу после рождения и является врожденной патологией.
Таким образом, не все врожденные заболевания наследственные и есть часть наследственных болезней, не являющихся врожденными. Фенокопия – ненаследственное изменение фенотипа организма, вызванное факторами окружающей среды и копирующее проявление какого-либо известного наследственного изменения (заболевания). Причиной фенокопии служит нарушение обычного хода индивидуального развития без изменения генотипа.
Генокопия - возникновение внешне сходных фенотипических признаков (заболеваний) под воздействием генов, расположенных в различных участках хромосомы или в различных хромосомах, т.е. заболевание предопределяется разными генами. Например, слепота может быть связана с генетическим поражением и сетчатки и хрусталика, которые контролируются различными генами. Существует несколько генокопий синдрома Дауна.
Врожденная патология, вызванная нарушениями развития плода, наблюдается у приблизительно 2% новорожденных и является наиболее частой причиной неонатальной смертности и заболеваемости. При большинстве аномалий не обнаруживается никаких хромосомных нарушений, и они не являются наследственными.
Среди наследственных болезней выделяют болезни с наследственной предрасположенностью (мультифакториальные). Наследственная предрасположенность подразумевает, что болезнь не детерминируется жестко генетическим аппаратом, но по наследству передаются некие свойства и особенности организма, его органов и систем, которые предрасполагают к возникновению определенных болезней (атеросклероз, гипертоническая болезнь, сахарный диабет, опухоли и др.). В основе мультифакториальных болезней лежит полигенное наследование, когда многие пары генов суммируют свое влияние (аддитивное действие)
Тератогенные факторы (или тератогены) - факторы, вызывающие пороки развития (от греч. teratos - уродство). Врожденные заболевания подразделяются в зависимости от срока возникновения. 1) период прогенеза соответствует созреванию гамет (яйцеклеток и сперматозоидов) до оплодотворения (в этот период возможно возникновение патологии гамет – гаметопатии) ; 2) период киматогенеза (от греч. kyema - зародыш) соответствует периоду от оплодотворения до родов. С периодом киматогенеза совпадает период киматопатии. В нем различают три периода:
· ранний фетальный период (76-180 день беременности) - возможно возникновение болезней раннего фетогенеза;
· поздний фетальный период (181-280 день беременности) - возможно возникновение болезней позднего фетогенеза. I. Методы изучения наследственных болезней.
Клинико-генеалогический метод заключается в составлении родословной записи с последующим анализом проявления признака, характерного для конкретной наследственной болезни на протяжении возможно большего числа поколений родственников пациента. Признаками наследственных болезней, установленных с помощью «родословных», являются: 1) обнаружение болезни «по вертикали»: из поколения в поколение беспрерывно (при доминантном типе наследования) или с некоторыми перерывами (при рецессивном типе наследования); 2) менделевские соотношения между числом больных и здоровых сибсов (3:1; 1:1; 1:0); 3) большая частота заболевания среди родственников, чем среди неродственников. Близнецовый метод состоит в сопоставлении внутрипарной конкордантности (идентичности) одно- и двуяйцевых близнецов, живущих в разных и в одинаковых условиях, по анализируемому патологическому признаку. В среднем на каждые 100 одноплодных родов приходятся одни близнецовые (многоплодные); при этом однояйцевые близнецы рождаются реже, чем двуяйцевые, примерно в 3-4 раза. О наследственной природе патологии свидетельствует высокая конкордантность по анализируемому признаку однояйцевых близнецов, живущих в разных условиях, и, наоборот, низкая конкордантность двуяйцевых близнецов, особенно живущих в одинаковых условиях. Напротив, высокая конкордантность по какому-либо патологическому признаку одно- и двуяйцевых близнецов, живущих в одинаковой среде, явно говорит против наследственного происхождения данной патологии и, наоборот, подтверждает решающее значение в ее развитии экзогенных (внешних) факторов. Популяционно-статистический метод заключается в составлении родословных среди большой группы населения, в пределах области или целой страны, в исследовании генетических изолятов. Изолят - это группа людей, от 500 человек до нескольких тысяч, живущая изолированно от всего остального населения страны. Генетический изолят характеризуется тем, что браки заключаются только в его пределах, с высокой частотой эндогамных браков. Это ведет в конце концов к генной изоляции от остального народа страны. В результате происходит передача аномальных рецессивных генов из гетерозиготных в гомозиготные пары, что сопровождается увеличением числа наследственных болезней. Цитологический метод -установление генетического пола при исследовании клеток на наличие телец Барра. Когда в клетке присутствует две Х хромосомы (как у нормальной женщины), одна из них (тельце Барра) инактивируется и конденсируется на ядерной мембране. Отсутствие тельца Барра свидетельствует о наличии только одной Х хромосомы (у нормального мужчины (XY) и при синдроме Шершевского-Тернера (ХО)). Тельца Барра наиболее легко определяются в мазках многослойного эпителия, которые получают путем соскабливания буккальной слизистой оболочки.
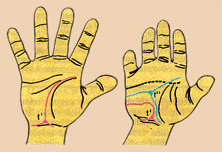
Биохимический и иммунологический методы заключаются в исследовании биохимических признаков, заведомо специфичных для определенных наследственных болезней. Так, например, для диагностики фенилпировиноградной олигофрении в моче определяют фенилпировиноградную кислоту; для диагностики серповидно-клеточной анемии (S-гемоглобиноза) исследуют наличие в крови S-гемоглобина; для выявления иммунодефицитных состояний определяют содержание различных антител и популяций лимфоцитов. Дерматоглифический метод – выявление наследственных болезней по рисунку ладоней.
Рис. 4.3. Схематическое изображение ладони здорового ребенка (слева) и ребенка того же возраста с болезнью Дауна. Цитогенетический метод состоит в микроскопическом исследовании структуры и числа хромосом клеток (лейкоцитов, эпителия и др.). Изменение структуры и числа хромосом (хромосомные аберрации) является признаком наследственной природы болезни.
Рис. 4.4. Схематическое изображение хромосом человека (идиограмма гаплоидного набора.
Рис. 4.5. Метафазная пластинка при простой окраске.
Молекулярно-генетический. Реализуется с помощью блот-гибритизации по Саузерну (введение флюоресцентной метки – ДНК-зонд) и амплификации (увеличении числа копий) участков ДНК при помощи ПЦР (полимеразной цепной реакции).
ПЦР осуществляется последовательными циклами. В каждом цикле происходят следующие события:
Затем процедуры повторяются сначала, и происходит копирование как старых, так и новых одноцепочечных цепей с образованием третьей и четвертой копий молекулы ДНК, затем все четыре снова копируются, и образуется уже восемь молекул ДНК, и т.д. число растет в геометрической прогрессии. В результате 20-30 циклов нарабатывается эффективное количество ДНК. Отдельный цикл занимает около 5 мин., а для бесклеточного молекулярного клонирования фрагмента ДНК требуется всего несколько часов. Метод ПЦР отличается очень высокой чувствительностью: он позволяет обнаружить в пробе всего одну присутствующую в ней молекулу ДНК. Тот же способ пригоден и для анализа следовых последовательностей РНК, для этого РНК переводят в последовательности комплементарной ДНК (кДНК), используя обратную транскриптазу. Метод получил широкое использование в пренатальной диагностике наследственных болезней, выявлении вирусных инфекций, а также в судебной медицине.
10. – Мутагены. Классификация мутаций. Генные и хромосомные болезни.
Мутация – скачкообразное изменение признака вследствие количественных или качественных изменений генотипа. Мутагены – факторы, вызывающие мутации. Мутации бывают : · соматические (потенциальное развитие опухолей) не передаются по наследству и, следовательно, не относятся к наследственным заболеваниям, хотя и поражают генетический аппарат клетки; · гаметические (передаются по наследству). § Летальные – сопровождаются гибелью организма внутриутробно, или сразу после рождения. § Сублетальные – гибель до полового созревания. § Гипогенитальные – сочетаются с бесплодием.
По характеру изменения генотипа в соответствии с тремя уровнями организации генетического материала (гены – хромосомы – геном) различают мутации – генные, хромосомные, геномные и цитоплазматические.
I. Генные мутации связаны с изменением структуры отдельных генов (участков ДНК, кодирующих синтез одного белка, одного признака). · Моногенная – мутация в одном гене с изменением одного признака (например, альбинизм, короткопалость); моногенные мутации обуславливают истинные наследственные заболевания. · Полигенные – одновременные мутации в различных генах различных хромосом, обуславливающие однонаправленные изменения в организме, которые определяют предрасположенность к некоторым заболеваниям (например, атеросклероз, гипертоническая болезнь, сахарный диабет II типа); наследуется не сама болезнь, а предрасположенность к ней, реализующаяся при воздействии определенных внешних факторов. Заболевание развивается как под влиянием мутаций, так и под влиянием факторов среды, т. е. является мультифакториальным. Даже для одного и того же заболевания относительное значение наследственности и среды у разных лиц может быть неодинаковым. · Точковая – повреждение одного нуклеотида в гене, т. е. замена одной аминокислоты в белке на другую (например, ферментопатии, серповидно-клеточная анемия, глухонемота).
II. Хромосомные мутации - структурные перестройки в отдельных хромосомах: делеции, дупликации, инверсии, транслокации. · Делеция - это потеря части хромосомы в результате ее разрыва. Большинство делеций летальны в результате потери огромной части генетического материала. Делеция короткого плеча 4 хромосомы приводит к развитию синдрома Вольфа; делеция короткого плеча 5 хромосомы - синдрома кошачьего крика (сri du chat) - мяуканье и звуки подобные кошачьему крику типичны для этой патологии, часто наблюдается отставание в умственном развитии и пороки сердца. · Транслокация - это перенос отдельного сегмента одной хромосомы в другую хромосому. При сбаласнсированной транслокации весь генетический материал сохраняется и остается фунционально способным, поэтому фенотипических проявлений нет. У таких людей могут формироваться аномальные гаметы. · Дупликация – удвоение участка хромосомы. · Инверсии – поворот участка хромосомы на 1800.
Рис. 4.6. Схематическое изображение различных видов хромосомных мутаций.
III. Геномные мутации - изменения числа хромосом в наборе, не сопровождаемые изменением их структуры. Число хромосом при этом меняется некратно – формируется анеуплоидный набор хромосом. Кратное изменение числа хромосом (полиплоидия) несовместимо с жизнью.
1. Моносомии – уменьшение количества хромосом. · Синдром Шерешевского-Тернера (яичниковая дисгенезия) - моносомия половых хромосом, встречается довольно часто. Отсутствует одна Х-хромосома (45, ХО). В некоторых случаях вторая Х-хромосома присутствует, но в ней выявляются тяжелые нарушения (изохромосома, частичная делеция и др.). Потеря второй Х-хромосомы обычно приводит к гибели плода. У выживших детей наблюдается лимфэдема шеи, которая присутствует и у взрослых, приводя к формированию толстой шеи. Часто наблюдаются врожденные аномалии сердца, низкий рост, ожирение и нарушения строения скелета. Интеллект не нарушен. В присутствии одной Х-хромосомы (и отсутствии Y-хромосомы) примитивные половые железы развиваются как яичники. Отсутствие второй Х-хромосомы приводит к нарушению развития яичников в пубертатном периоде. Яичники остаются маленькими и в них не обнаруживаются примордиальные фолликулы. Также нарушается синтез эстрогенов, что проявляется нарушением эндометриального цикла (аменоррея) и слабым развитием женских вторичных половых признаков. Диагноз может быть поставлен при отсутствии телец Барра в соскобах буккального эпителия у лиц, имеющих женский фенотип или при анализе кариотипа.
· При аутосомной моносомии теряется огромное количество генетического материала, поэтому она обычно летальна.
2. Триосомии – увеличение количества хромосом на одну. · Синдром Кляйнфельтера (тестикулярная дисгенезия) - трисомия половых хромосом - встречается довольно часто. Проявляется наличием лишней Х-хромосомы (47, ХХY), реже больные с синдромом Кляйнфельтера могут иметь две и более лишних Х-хромосом (48, ХХХY или 49, ХХХХY). Формируется мужской фенотип. До пубертатного периода никаких клинических проявлений не наблюдается. Лишняя Х-хромосома нарушает нормальное развитие яичек в пубертатном периоде неизвестным способом. Яички остаются маленькими и не продуцируют сперматозоиды, больные обычно бесплодны. Уровень тестостерона в крови низкий, что приводит к нарушению развития вторичных половых признаков. У больных имеется склонность к высокому росту (тестостерон ускоряет окостенение эпифизов) и евнухоидный внешний вид с высоким голосом, маленьким пенисом и ростом волос по женскому типу. Также иногда наблюдается гинекомастия. Иногда наблюдается снижение интеллекта. Диагноз синдрома Кляйнфельтера может быть установлен при нахождении телец Барра в соскобах буккального эпителия у лиц, имеющих мужской фенотип или при анализе кариотипа. · Синдром ХХХ (“суперженщины”) - присутствие третьей Х-хромосомы у женщин. Большинство пациентов являются нормальными. У некоторых наблюдаются нарушения умственного развития, нарушения менструального цикла и снижение фертильности (плодовитости). · Синдром ХYY - присутствие лишней Y-хромосомы у мужчин. Большинство пациентов нормальные. У некоторых может наблюдаться агрессивность поведения и легкое отставание в умственном развитии. · Синдром Дауна является наиболее частым аутосомным нарушением. Он возникает в результате наличия третьей 21 хромосомы, что приводит к развитию характерных клинических проявлений. Дети имеют характерный косой разрез глаз с уплощенным профилем, кососмотрящие глаза, резко выраженные вертикальные кожные складки, прикрывающие медиальный угол глазной щели, так называемое сходство с лицами азиатов, раньше называемое “монголоидным”. Постоянным признаком является отставание в умственном развитии. 30% пациентов имеют врожденные пороки сердца. Также у этих больных повышена заболеваемость различными инфекциями, язвами двенадцатиперстной кишки и острой лейкемией. Мужчины с синдромом Дауна обычно бесплодны, а женщины могут рожать детей. Потомки матерей с синдромом Дауна могут быть нормальными, потому что лишняя 21 хромосома содержится не во всех гаметах.
Рис. 4.7. Дети с синдромом Дауна.
· Синдром Эдвардса - трисомия 18 хромосомы (47ХХ/ХY, +18) встречается редко. Клинически он проявляется отставанием в физическом и умственном развитии, сопровождаемым характерными физическими недостатками, такими как “стопа рокера” и сжатые в кулаки руки с перекрещивающимися пальцами. В результате тяжелых поражений дети редко выживают более года. · Синдром Патау - трисомия 13 хромосомы (47ХХ/ХY, +13) также встречается редко. Большинство детей умирает сразу после рождения. Трисомия 13 хромосомы характеризуется нарушением развития подкорковых структур мозга (отсутствие обонятельных луковиц, слияние лобных долей и единственный желудочек головного мозга) и срединных структур лица (расщепление губы, расщепление твердого неба, дефекты носа, единственный глаз [циклоп]).
IV. Цитоплазматические (митохондриальные) мутации возникают в результате мутаций в плазмогенах, находящихся в ДНК-содержащих клеточных органеллах - митохондриях. Некоторые патологии, связанные приводящие к мужскому бесплодию, связаны с этим видом мутаций. Некоторые виды близнецовости могут быть обусловлены этими же причинами, при этом наследуются, как правило, только по женской линии.
В стандартной родословной используются простые условные обозначения и правила:
11. – Нарушение регионального кровообрашения. Механизмы артериальной и венозной гиперемии, ишемии и стаза.
Региональное кровообращение (периферическое, органное) – система, обеспечивающая циркуляцию крови в органах и тканях большого круга кровообращения.
|
||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-06-19; просмотров: 730; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.145.63.136 (0.081 с.) |