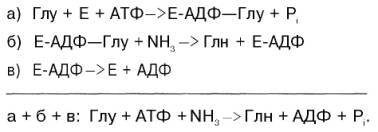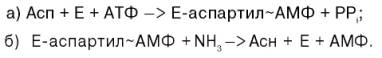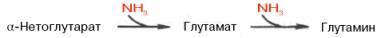Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Биосинтез и катаболизм пиримидиновых нуклеотидов. Регуляция синтеза.
1. УМФ является общим предшественником всех остальных пиримидиновых нуклеотидов: ЦМФ и ТМФ. Синтез У М Ф de novo включает шесть последовательных стадий (рис. 10.5) и протекает главным образом в цитозоле клеток при участии трех ферментов, два из которых полифункциональны: • первый полифункциональный фермент - КАД-фермент содержит домены, проявляющие активности карбамоилфосфатсинтетазы II (КФСН), аспартаттранскарбамоилазы (ATK), дигидрооротазы и катализирующие три первые реакции этого метаболического пути; • митохондриальная NAD-зависимая дигидрооротатдегидрогеназа которая окисляет дигидрооротат в оротат; • превращение азотистого основания - оротата в нуклеотид и его последующее декарбоксилирование до УМФ катализирует второй полифункциональный фермент - УМФ-синтаза, обладающая оротатфосфорибозилтрансферазной и ОМФ-декарбоксилазной активностями.
1-3 - КАД-фермент (1-карбамоилфосфатсинтетаза, КФС11); 2 - аспартаттранскарбамоилаза (АТК); 3 -дигидрооротаза; 4 - дигидрооротатдегидрогеназа; 5, 6 - УМФ-синтаза (5-оротатфосфорибозилтрансфераза; 6 - ОМФ-декарбоксилаза); 7 - НМФ-киназа; 8 - НДФ-киназа; 9 - ЦТФ-синтетаза. Аллостерически регулируются по механизму отрицательной обратной связи карбамоилфосфатсинтетазная и аспататтранскарбамоилазная активности КАД-фермента конечными продуктами метаболического пути - УМФ и ЦТФ, тогда как ФРДФ активизирует КФС11, а АТФ - АТК Пиримидиновое кольцо «собирается» из простых предшественников - глутамина, аспартата и СО2 - и после окисления превращается в азотистое основание - оротат. Превращение оротата в нуклеотид - оротидин-5'- монофосфат (ОМФ) - осуществляется с использованием ФРДФ. УМФ образуется при декарбоксилировании ОМФ. Превращение УМФ в полифосфорные производные за счет переноса γ-фосфатного остатка АТФ на УМФ с образованием УДФ и УТФ катализируют НМФ- и НДФ-киназы. Синтез ЦТФ из УТФ осуществляет ЦТФ-синтетаза, используя амидную группу Глн и энергию АТФ для аминирования пиримидинового кольца. 2. Регуляция процесса. Активность ферментов синтеза пиримидиновых нуклеотидов регулируется аллостерически по механизму отрицательной обратной связи конечными продуктами УМФ и ЦТФ:
• в составе КАД-фермента: УМФ ингибирует, а ФРДФ активирует КФС II, тогда как активность аспартаттранскарбамоилазы ингибирует ЦТФ, но активирует АТФ; • УМФ и ЦМФ снижают активность второго полифункционального фермента - УМФ-синтазы; • накопление ЦТФ снижает активность ЦТФ-синтетазы. Синтез пуриновых и пиримидиновых нуклеотидов строго координируется: ФРДФ активирует оба синтеза, а накопление пуриновых и пиримидиновых нуклеотидов ингибирует образование ФРДФ по механизму отрицательной обратной связи.
5. Катаболизм пиримидиновых нуклеотидов. От пиримидиновых нуклеотидов отщепляются остатки фосфорной кислоты и рибозы в реакциях, аналогичных катаболизму пуриновых нуклеотидов. Образующиеся пиримидиновые основания способны разрушаться ферментными системами организма с образованием ряда продуктов (рис. 10.6).
В отличие от конечных продуктов пуринового обмена продукты катаболизма пиримидинов хорошо растворимы в воде. Причем β-аланин имеет физиологическое значение, содержится во многих тканях и в плазме крови в свободном виде или включается в состав мышечных пептидов: карнозина и ансерина. Бактериальные клетки используют β-аланин для синтеза пантотеновой кислоты, которая входит в состав HS-КоА.
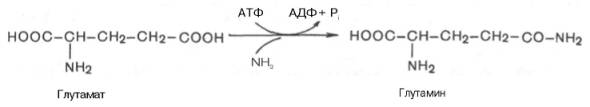
β-Аминоизобутират, образующийся при распаде тимина, частично экскретируется с мочой, а частично трансаминируется, превращаясь в метилмалонил полуальдегид, а затем в сукцинил-КоА. заболеваниями. 86.Реакции образования и обезвреживания аммиака в организме человека. Причины гипераммониемии. В организме человека подвергается распаду около 70 г аминокислот в сутки, при этом в результате реакцийдезаминирования и окисления биогенных аминов освобождается большое количество аммиака, являющегося высокотоксичным соединением. Поэтому концентрация аммиака в организме должна сохраняться на низком уровне. Действительно, уровень аммиака в крови в норме не превышает 60 мкмоль/л (это почти в 100 раз меньше концентрации глюкозы в крови). В опытах на кроликах показано, чтоконцентрация аммиака 3 ммоль/л является летальной. Таким образом, аммиак должен подвергаться связыванию в тканях с образованием нетоксичных соединений, легко выделяющихся с мочой. Один из путей связывания и обезвреживания аммиака в организме, в частности в мозге, сетчатке, почках,печени и мышцах,– это биосинтез глутамина (и, возможно, аспарагина). Глутамин и аспарагин выделяются смочой в небольшом количестве. Было высказано предположение, что они выполняют скорее транспортную функцию переноса аммиака в нетоксичной форме. Ниже приводится химическая реакция синтеза глутамина, катализируемого глутаминсинтетазой.
Механизм этой синтетазной реакции, подробно изученный А. Майсте-ром, включает ряд стадий. Синтезглутамина в присутствии глутамин-синтетазы может быть представлен в следующем виде:
Биосинтез аспарагина протекает несколько отлично и зависит от природы ферментов и донора аммиака. Так, у микроорганизмов и в животных тканях открыта специфическая аммиакзависимая аспарагинсинтетаза, которая катализирует синтез аспарагина в две стадии:
В животных тканях содержится, кроме того, глутаминзависимая аспа-рагинсинтетаза, которая для синтеза во второй стадии использует амидную группу глутамина: б) Е-аспартил~АМФ + Глн -> Асн + Е + АМФ + Глу. Суммарная ферментативная реакция синтеза аспарагина может быть представлена в следующем виде: Асп + АТФ + NН3 (или Глн) –> Асн + АМФ + РРi + (Глу). Видно, что энергетически синтез аспарагина обходится организму дороже, поскольку образовавшийся РРiдалее распадается на ортофосфат. Часть аммиака легко связывается с α-кетоглутаровой кислотой благодаря обратимости глутаматдегидрогеназной реакции. Если учесть связывание одной молекулы аммиака при синтезе глутамина, то нетрудно видеть, что в организме имеется хорошо функционирующая система, связывающая двемолекулы аммиака:
Глутамин, кроме того, используется почками в качестве резервного источника аммиака (образуется изглутамина под действием глутаминазы), необходимого для нейтрализации кислых продуктов обмена при ацидозе и защищающего тем самым организм от потери с мочой используемых для этих целей ионов Na+.
Гипераммониемия — это нарушение обмена веществ, проявляющееся в недостаточности цикла ферментов мочевины, приводящее к отравлению организмааммиаком. Аммиак является токсичным соединением, находящимся в крови в относительно небольших концентрациях (11,0—32,0 мкмоль/л). Симптомы аммиачного отравления проявляются при превышении этих пределов всего в 2—3 раза. Предельно допустимый уровень аммиака в крови 60 мкмоль/л. При повышении концентрации аммиака (гипераммониемия) до предельных величин может наступить кома и смерть. При хронической гипераммониемии развивается умственная отсталость. Транзиторной гипераммониемией называется также пограничное состояние, присущее новорожденным детям в период адаптации к внеутробной жизни, проявляющееся обычно на вторые—третьи сутки жизни. Этот вид гипераммониемии встречается чаще всего у недоношенных детей с задержкой внутриутробного развития, с частотой до пятидесяти процентов рождений, однако иногда регистрируется и у доношенных малышей. Часть детей не проявляет симптоматики клинической картины гипераммониемии: признаки угнетения центральной нервной системы (вялость, понижение мышечного тонуса, приступы апноэ, ослабленная реакция зрачков на свет, отказ от еды, ступор и кома), а также расстройства дыхательной функции, желтуха, судороги и обезвоживание. Причиной, вызывающей гипераммониемию, называют кислородное голодание, или гипоксию, во время беременности и в процессе родов. Приобретённые формы Приобретённая (вторичная) гипераммониемия развивается вследствие заболеваний печени и вирусных инфекций. В крайне тяжёлых случаях она проявляется как тошнота, рвота, судороги, нечленораздельная речь, затуманивание зрения, тремор, нарушение координации движений. Наследственные формы Наследственные формы гипераммониемии вызваны генетическим дефектом любого из пяти ферментов синтеза мочевины. Соответственно ферменту заболевание делится на пять типов. Первичными признаками гипераммониемий являются сонливость, отказ от пищи, рвота, беспокойство, судороги, нарушение координации движений, тахипноэ, дыхательный алкалоз. Могут развиться печёночная недостаточность, лёгочные и внутричерепные кровоизлияния. Наиболее частой является гипераммониемия типа II, связанная с недостатком орнитин-карбамоилтрансферазы. Заболевание рецессивно, сцеплено с Х-хромосомой. У матери также наблюдается гипераммониемия и отвращение к белковым продуктам. При полном дефекте фермента наследственные гипераммониемии имеют раннее начало (в период до 48 часов после рождения).
Лабораторным критерием заболевания является накопление глутамина (в 20 и более раз) и аммиака в крови, ликворе и моче. Основа лечения гипераммониемий сводится к ограничению белка в диете, уже это позволяет предотвратить многие нарушения мозговой деятельности. Причины Гипераммониемии: Токсичность аммиака обусловлена следующими обстоятельствами: 1. Связывание аммиака при синтезе глутамата вызывает отток α-кетоглутарата из цикла трикарбоновых кислот, при этом понижается образование энергии АТФ и ухудшается деятельность клеток. 2. Ионы аммония NH4+ вызывают защелачивание плазмы крови. При этом повышается сродство гемоглобина к кислороду (эффект Бора), гемоглобин не отдает кислород в капиллярах, в результате наступает гипоксия клеток. 3. Накопление свободного иона NH4+ в цитозоле влияет на мембранный потенциал и работу внутриклеточных ферментов — он конкурирует с ионными насосами для Na+ и K+. 4. Продукт связывания аммиака с глутаминовой кислотой — глутамин — является осмотически активным веществом. Это приводит к задержке воды в клетках и их набуханию, что вызывает отёк тканей. В случае нервной ткани это может вызвать отёк мозга, кому и смерть.
|
|||||||||||
|
|
Последнее изменение этой страницы: 2021-07-18; просмотров: 605; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.161.116 (0.011 с.) |
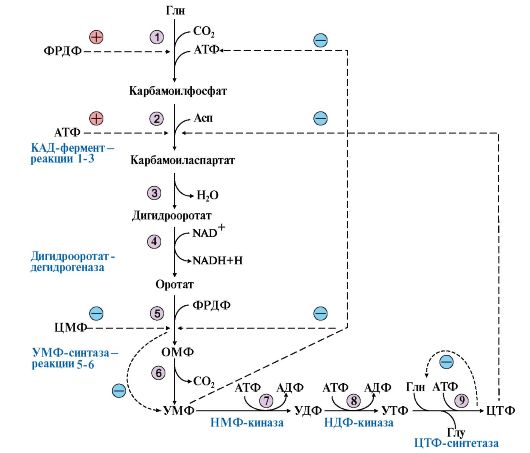 Рис. 10.5. Синтез пиримидиновых нуклеотидов и его регуляция:
Рис. 10.5. Синтез пиримидиновых нуклеотидов и его регуляция: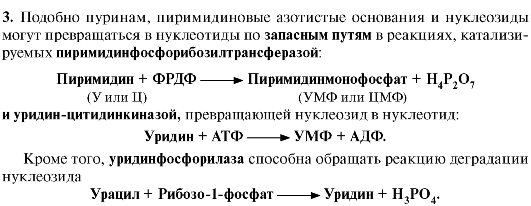 4. Оротацидурия - редкое наследственное заболевание, при котором в результате мутации в гене второго полифункционального фермента нарушается превращение оротата в УМФ. С мочой выделяется до 1,5 г оротата (в 1000 раз больше, чем в норме). Клинически заболевание проявляется в виде мегалобластной анемии, нарушений работы ЖКТ, сердца, интеллектуального развития и двигательной способности. Высокие концентрации оротата в крови и тканях нетоксичны для организма, а указанные симптомы возникают в результате неспособности организма из-за «пиримидинового голода» обеспечить нормальную скорость синтеза нуклеиновых кислот и увеличения числа клеток. Наиболее характерным следствием оротацидурии является мегалобластная анемия, вызванная снижением скорости деления клеток эритроцитарного ряда. Для лечения этой болезни используют уридин или цитидин в дозах от 0,5 до 1 г/сут, которые с помощью уридинцитидинкиназы превращаются в УМФ или ЦМФ в обход нарушенной реакции. Полифосфорные производные полученных нуклеотидов - УТФ и ЦТФ - ингибируют регуляторные ферменты синтеза пиримидиновых нуклеотидов de novo и снижают синтез и выведение оротата с мочой.
4. Оротацидурия - редкое наследственное заболевание, при котором в результате мутации в гене второго полифункционального фермента нарушается превращение оротата в УМФ. С мочой выделяется до 1,5 г оротата (в 1000 раз больше, чем в норме). Клинически заболевание проявляется в виде мегалобластной анемии, нарушений работы ЖКТ, сердца, интеллектуального развития и двигательной способности. Высокие концентрации оротата в крови и тканях нетоксичны для организма, а указанные симптомы возникают в результате неспособности организма из-за «пиримидинового голода» обеспечить нормальную скорость синтеза нуклеиновых кислот и увеличения числа клеток. Наиболее характерным следствием оротацидурии является мегалобластная анемия, вызванная снижением скорости деления клеток эритроцитарного ряда. Для лечения этой болезни используют уридин или цитидин в дозах от 0,5 до 1 г/сут, которые с помощью уридинцитидинкиназы превращаются в УМФ или ЦМФ в обход нарушенной реакции. Полифосфорные производные полученных нуклеотидов - УТФ и ЦТФ - ингибируют регуляторные ферменты синтеза пиримидиновых нуклеотидов de novo и снижают синтез и выведение оротата с мочой.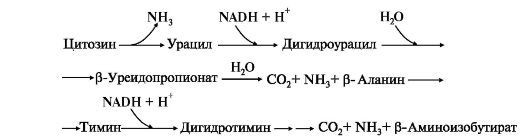 Рис. 10.6. Катаболизм пиримидиновых оснований
Рис. 10.6. Катаболизм пиримидиновых оснований