
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Основные представления химической кинетики
Энергия активации химических реакций. Почему химические реакции, идущие с образованием энергетически более бедных соединений, протекают не самопроизвольно, а только после соответствующего возбуждения или в присутствии катализаторов? Например, при обычных условиях водород и кислород Н2 и О2 самопроизвольно не образуют воду, а азот и водород – аммиак. Ответы на такие вопросы связаны не только с фундаментальными проблемами химии, но и всей живой природы. Если бы все в природе определялось только энергией, то в природе могли бы существовать лишь простейшие соединения, которые энергетически наиболее стабильны. Законы химической термодинамики определяют возможность, направление и степень протекания химического превращения веществ – химической реакции. Эти сведения получают путем расчета величины изменения свободной энергии Однако условие
Сказанное можно проиллюстрировать примером из механики. Имеются три тела с одинаковой массой m, но различной формы (рисунок 117) с центрами тяжести S, распо-ложенными на различной высоте от общей опорной плоскости.
Рисунок 117
Все три тела можно опрокинуть так, что их центр тяжести займет наинизшее положение. С точки зрения законов физики, наиболее стабильным будет состояние тела, обладающего наименьшей потенциальной энергией U, центр тяжести которого расположен ниже, чем у других. Потенциальная энергия этих трех тел увеличивается в следующей последовательности
где С энергетических позиций, самым стабильным должно быть тело 1. Однако из рисунка видно, что его положение неустойчиво и при малейшем внешнем воздействии оно упадет (если нам удастся поставить его в это положение). Потенциальная энергия тела 2 больше, чем тела 1, но его положение более устойчиво, и чтобы привести его в наклонное положение, надо сначала поднять центр тяжести на высоту Таким образом, для того чтобы опрокинуть тела 2 и 3, вначале их нужно активировать, затратив на это определенную энергию активации
В качестве химического процесса рассмотрим образование соляной кислоты из водорода и хлора
В данном процессе выделяется 184 кДж энергии, поэтому 2 моль HСl стабильнее, чем смесь 1 моль газообразного водорода Н2 и 1 моль газообразного хлора Cl2. Несмотря на это, при обычной температуре или в темноте этот процесс не идет. Образование конечных продуктов химической реакции связано с перегруппировкой атомов и с изменением химических связей между ними. В исходных веществах (молекулах Н2 и Cl2) химически связанные атомы должны отделиться друг от друга, чтобы затем соединиться по-другому и образовать продукт реакции (в нашем случае соляную кислоту). Чтобы реакция пошла, необходимо вначале разорвать химические связи в Н2 и Cl2, но для этого требуется энергия активации. Если мы примем, что для образования соляной кислоты необходимо разложить молекулу Cl2 на атомы, то потребуется 239 кДж энергии, в случае молекулы Н2 эта величина существенно выше – 431 кДж. Свободные атомы Cl могут так располагаться вокруг молекулы Н2, что одновременно с образованием связей Н – Cl будут разрываться связи Н – Н и при этом образуются молекулы HСl. На рисунке 118 схематически представлены эти энергетические соотношения. Для приведения системы в активированное состояние, имеющее энергию 910 кДж (Н2+2Cl), необходимо к химической энергии (энергии связи) в 672 кДж, соответствующей начальному состоянию Н2+Cl2, добавить из внешнего источника еще 238 кДж. Из активированного состояния система переходит в конечное состояние 2НCl, которому соответствует энергия 488 кДж. Во время перехода выделяется обратно 238 кДж подведенной энергии активации и 184 кДж – в виде теплоты. Эта последняя величина и есть разница энергий между конечным состоянием 2НCl и начальным состоянием Н2 + Cl2. Возникает вопрос, откуда может быть получена энергия, необходимая для активации процесса образования НСl. Активацию можно осуществить: 1. Кинетической энергией соударяющихся частиц. Если сталкивающиеся молекулы, имеют достаточно высокую энергию, то при этом может произойти разрыв химических связей и появится возможность перегруппировки освободившихся атомов с образованием молекулы с меньшей энергией. В темноте и при комнатной температуре хлор и водород не вступает в реакцию с образованием соляной кислоты. Это свидетельствует о том, что энергия, выделяющаяся при столкновениях молекул меньше энергии активации. Но при высоких температурах (несколько сот градусов) реакция протекает очень бурно и даже со взрывом.
Проблему проясняет молекулярно-кинетическая теория вещества. Взаимодействуя (сталкиваясь) между собой, частицы обмениваются энергией, и в системе, находящейся в тепловом равновесии, устанавливается некоторое стационарное распределение энергии (скорости) между всеми молекулами. Такое состояние совсем не означает, что все молекулы обладают одинаковой энергией. Это невозможно в динамической системе. Распределение является статистическим. Большинство молекул имеет энергии, близкие к средней, а число молекул с меньшими и большими энергиями уменьшается. На рисунке 119 приведено распределение по энергии молекул идеального газа при различных температурах, так называемое распределение Максвелла–Больцмана:
где dN – число молекул, имеющих энергию в интервале от Е до Е + dE; N – полное число молекул в системе. Чтобы определить число молекул с энергиями, большими энергии активации
С повышением температуры доля активных молекул и, следовательно, скорость реак-ции быстро возрастает. При еще более высокой температуре почти в каждом столкно-вении выделяется энергия, достаточная для активации химического превращения. Именно такое состояние возникает в реакции образования соляной кислоты, когда при высокой температуре реакция протекает со взрывом. 2. Активация может происходить также под воздействием света. Если фотон с длиной волны меньше 550 нм поглощается молекулой Cl2, то она диссоциирует на два атома
Образовавшиеся при этом атомы начинают цепную реакцию
и хлорводородный гремучий газ взорвется даже при комнатной температуре – с большой скоростью превратится в соляную кислоту. Кинетический метод управления химическими процессами. Химическое превра-щение протекает во времени, которое не фигурирует в термодинамическом описании, рассмотренном выше. В основе химической реакции лежат физические процессы переме-щения атомов – переходы от одной молекулярной структуры к другой, изменения их электронных состояний. Важнейшей характеристикой химических реакций является скорость химических реакций.
Она определяется количеством превращенного (исчезнувшего или образовавшегося) вещества Для гетерогенной системы, в которой реакция происходит на границе раздела фаз с площадью S, Количество вещества, отнесенное к занимаемому объему есть молярная концентрация. Поэтому скорость гомогенной реакции можно определить как изменение концентрации одного из реагирующих веществ (исходных или конечных) в единицу времени
где С – концентрация. Если С – концентрация исходного вещества, то Основные законы химической кинетики. Направление химического процесса определяется не только химической природой веществ, но и их концентрацией. В 1879 г. К. Гульдберг и П. Вааге предложили закон действующих масс, согласно ко-торому скорость реакции определяется вероятностью столкновения реагирующих частиц, то есть прямо пропорциональна их концентрации. Скорость реакции определяется числом столкновений и пропорциональна их вероятности. Если рассматривать реакцию между А и В, концентрация которых СА и СВ, то вероятность нахождения частицы в какой-либо точке пространства ~ СА, а второго СВ. При условии независимого движения обоих сортов атомов вероятность их нахождения в данных точках определяется произведением САСВ. Обозначив коэффициент пропорциональности k, получим:
где k – называется константой скорости реакции. Уравнения, устанавливающие зависимость скорости реакции от концентрации, называются кинетическими. Из приведенных кинетических уравнений видно, что размерность константы скорости зависит от суммы показателей степеней при концентрациях веществ в этих уравнениях. Эта сумма называется порядком реакции. Реакция (1) – реакция второго порядка – размерность k с учетом размерности Для управления химическим процессом важнейшее значение имеет температурная зависимость скорости реакции. Так как, скорость реакции зависит от концентрации взаимодействующих веществ, то для исследования температурной зависимости скорости нужно измерять ее при одинаковой концентрации веществ. Измеряя скорости различных химических реакций Аррениус (1889) установил, что константа скорости реакции k находится в экспоненциальной зависимости от абсолютной температуры Т
где Аррениус первым интерпретировал энергию активацию
Более наглядно температурную зависимость скорости реакции можно представить с помощью правила Вант-Гоффа, предложенного им в 1884 г.:
где Согласно правилу Вант-Гоффа, с повышением температуры на 10 оС скорость ре-акции возрастает в 2–4 раза, т.е. коэффициент Катализ. Известно, что многочисленные химические реакции могут быть в значительной степени ускорены только благодаря присутствию определенных веществ. Это явление называется катализом, а действующее вещество – катализатором. Катализаторы в реакциях не расходуются и в состав конечных продуктов не входят. Например, в смеси кислорода и водорода (гремучий газ) при комнатной температуре в течение десятилетий не наступает никаких самопроизвольных изменений. Но если привести эту газовую смесь в соприкосновение с мелкодисперсной платиной, то водород и кислород мгновенно взрываются с образованием воды. При этом катализатор платина остается практически неизменным. Существует очень большое количество катализаторов, которые играют исключительно важную роль в регулировании процессов, происходящих в неживой и живой природе. Особенно активными катализаторами являются ферменты, которые обеспечивают протекание химических реакций при невысоких температурах живых организмов. Удивляет каталитическая активность ферментов, которые увеличивают скорость биохимических реакций в тысячу и десятки тысяч раз. Например, 1 моль алкоголь дегидрогеназы за 1 с при комнатной температуре превращает в уксусный альдегид 720 моль этанола. В то время как промышленные катализаторы того же процесса (медь) при 200 оС за 1 с превращают 0,1–1,0 моль спирта на 1 моль катализатора. Вещества, замедляющие реакции, называются ингибиторами. Катализ принято подразделять на следующие типы: а) гомогенный, когда реакционная смесь и катализатор находятся или в жидком, или в газообразном состоянии; б) гетерогенный, когда катализатор находится в виде твердого вещества, а реагирующие соединения в жидкой или газообразной смеси; в) ферментативный, когда катализатором служат сложные белковые образования, которые могут быть как гомогенными, так и гетерогенными. Примером гомогенного катализа в газовой фазе служит реакция окисления оксида углерода в присутствии паров воды. В гомогенных системах реакция идет во всем реакционном объеме При гетерогенном катализе реакция происходит на поверхности раздела фаз, причем решающую роль играет строение поверхности твердого вещества – катализатора. Она должна быть достаточной большой, чтобы обеспечивать максимальную величину реак-ционной зоны. Поэтому твердый катализатор используется в мелкодисперсном состоянии. После открытия Берцелиусом катализаторов в 1836 году в течение долгого времени оставалось загадкой, каким образом они осуществляют свое ускоряющее действие, не участвуя сами в химическом процессе. Дальнейшие исследования показали, что катализаторы все же временно очень активно участвуют в химическом процессе, но по окончании своего воздействия снова превращаются в исходное состояние. После чего они вновь готовы содействовать превращению молекул. Вышеизложенное справедливо, если катализатор в результате реакций не изменяется. На самом деле, особенно в случае гетерогенного катализа, он, хотя и незначительно, меняется. Например, может измениться его дисперсность, что влечет за собой уменьшение активной поверхности. Может измениться строение этой поверхности или ее химический состав за счет поглощения некоторых веществ из окружающей среды, что сопровождается отравлением катализатора. Обычно вклад таких изменений в общую энергетику химического процесса пренебрежимо мал и практически его не учитывают. Опытным путем удалось установить, что катализаторы уменьшают энергию активации процесса и тем самым ускоряют его. Например, энергия активации разложения перекиси водорода ( Взаимодействия между катализатором и реагирующим веществом весьма разно-образны и еще недостаточно изучены. Однако можно выделить два основополагающих аспекта. Во-первых, каталитическое действие твердых катализаторов основано главным образом на том, что они связывают молекулы исходных веществ на своей поверхности (адсорбируют их). При этом молекулы так деформируются, что ослабляются те связи, разрыв которых необходим для химической реакции. Во-вторых, энергия связи атомов реагирующей группировки с поверхностными атомами должна быть не слишком слабой и не слишком прочной, ибо в первом случае не произойдет необходимого ослабления связей в реакционной группе, а во втором – не произойдет десорбции продуктов реакции с поверхности катализатора и последняя будет отравлена. По такому механизму происходит реакция водорода Н2 с кислородом О2 под воздей-ствием платины. Опытным путем установлено, что молекулярный водород растворяется в платине, при этом его молекулы распадаются на свободные атомы ( Направляющее действие катализатора. Роль катализатора не сводится только к ускорению реакции. Большинство химических реакций проходит через несколько стадий, в реакционной смеси одновременно идет несколько реакций. Катализатор, снижая при 250 оС на окиси алюминия образуется уксусный альдегид
при 350 оС на той же окиси алюминия получается этилен
при 200 оС на активной меди образуется уксусная кислота
Иногда каталитическое действие оказывает один из продуктов реакции. Такие реакции называются автокаталитическими, а само явление автокатализом. 11 Эволюционная химия
Четвертый концептуальный уровень развития химических знаний связан с возникновением во второй половине ХХ в. эволюционной химии. Эволюционная химия включает учение об открытых высокоорганизованных химических системах, эволюция которых связана с ферментативным катализом. В основе эволюционной химии лежат принципы и законы, на которых построена биохимия живых организмов. По современным представлениям живые организмы представляют собой открытые неравновесные самоорганизующиеся системы. Химики надеются в рамках самоорганизующихся систем осуществить синтез новых сложных веществ с заданными свойствами без участия человека. Изучение биохимических процессов, протекающих в живой клетке, может быть использовано для создания принципиально нового управления химическими реакциями. Химический реактор на уровне эволюционной химии представляет собой некое подобие живой системы, для которой характерны саморазвитие и высокая степень организации. Первым ученым, обратившим внимание на высокую упорядоченность и эффективность химических процессов в живых организмах, был основатель органической химии Берцелиус. Он установил, что основой клеточного биохимического реактора является биокатализ. Идеалом совершенства каталитических превращений считали лабораторию живого организма Либих, Бертло и другие. Мы уже не раз упоминали слово «фермент». Практически ни один процесс в биологической системе не проходит без участия ферментов. Биохимию в целом можно определить как химию ферментативных реакций. Ферментов известно великое множество. Так, одна-единственная клетка простейшей бактерии использует в своей жизнедеятельности около тысячи разных ферментов. Ферменты катализируют тысячи реакций, идущих в живой клетке, при дыхании, обмене веществ, размножении и др. И самое замечательное свойство ферментов – работают они чрезвычайно быстро. Чтобы расщепить какой-либо белок или молекулу полиуглевода (крахмал, целлюлоза) на составные части, их нужно кипятить с крепкими растворами кислот и щелочей несколько часов. Ферменты пищеварительных соков – пепсин, протеаза, амилаза – гидролизуют эти вещества за несколько секунд при температуре 37 оС. Ферменты – это биологические катализаторы. Как же действуют ферменты, почему они ускоряют в тысячи раз те или иные реакции? Каждый фермент – это молекула белка, свернутая в клубок – глобулу. Важнейшая часть такой глобулы – активный центр, небольшая область, в которой происходит реакция, управляемая и ускоряемая ферментом. Представим, что в активном центре молекулы фермента есть два углубления (кармана), в которые входят молекулы реагенты. Атомы активного центра приходят в быстрое движение, затем начинают двигаться (более медленно) другие части глобулы; они сжимают молекулы реагентов, как клещи, или растягивают ее, поворачивают, ломают. Концы молекул реагентов оказываются рядом – возникает новая химическая связь и образуется новая молекула. Она по своей конфигурации уже не подходит к активному центру в глобуле фермента, и фермент эту молекулу выталкивает. На освободившееся место встают две другие реагирующие молекулы. Но фермент может действовать не только механически, он может на какое-то время сам связываться с одной из реагирующих молекул. Такая молекула, после того как к ней присоединится фермент, обладает уже иными химическими свойствами, она гораздо активнее реагирует с другой молекулой. После этого фермент отщепляется. Показателен процесс расщепления ферментом лизоцимом молекулы полисахарида. Активный центр в молекуле лизоцима имеет форму щели, в которую укладывается длинная цепь полисахарида. При этом фермент изменяет свою конфигурацию, атомы, образующие щель, смещаются один относительного другого, и молекула полисахарида оказывается разрезанной на две половины, которые немедленно отделяются от фермента. Это, конечно, только схема. В действительности все гораздо сложнее, и еще многие детали биохимических процессов пока неясны. Изучая сложнейшие процессы, происходящие в живой клетке с участием ферментов, ученые задаются вопросом, а нельзя ли, поучившись у природы, провести в колбах и реакторах искусственные химические процессы, копирующие, моделирующие биохимические реакции. В настоящее время ученые научились создавать биокатализаторы, которые являются аналогами природных ферментов. Пока они несовершенны, они очень неустойчивы при хранении и быстро портятся, теряя свою активность. Систематические исследования ученых привели к созданию устойчивых, так называемых иммобилизованных ферментов (лат. immobilis – неподвижный). Сущность иммобилизации состоит в закреплении выделенных из живого организма ферментов на твердой поверхности путем адсорбции, которая превращает их в гетерогенный катализатор непрерывного действия. Основоположником химии иммобилизованных систем является советский химик И.В. Березин. Химические технологии, основанные на использовании иммобилизованных ферментов, реализованы в промышленных масштабах: в тонком органическом синтезе, трансформации стероидов, разделении рацематов на оптически активные формы. Изучаются возможности применения иммобилизованных систем для тяжелого органического синтеза, в частности, для получения на основе предельных и ароматических углеводородов: спиртов, альдегидов, кетонов, кислот. Большие надежды связываются с созданием принципиально новой химической технологии ферментативного обезвреживания сточных вод. Химическая эволюция и биогенез. В настоящее время нет общепринятой теории химической эволюции, приводящей к возникновению биологической формы движения материи – жизни. Наиболее последовательной является теория химической эволюции и биогенеза, предложенная в 1964 г. советским химиком А.П. Руденко. В основе этой теории лежит представление о том, что химическая эволюция тесно связана с саморазвитием каталитических систем. Среди разнообразных каталитических проявлений обычного типа были обнаружены катализаторы, способные к самосовершенствованию в ходе химической реакции. В этом случае происходит естественный отбор тех каталитических центров, которые обладают наибольшей активностью. Те же центры, изменение которых связано с уменьшением активности, постепенно исключаются из кинетического процесса. При многократных последовательных необратимых изменениях катализатора происходит переход его на более высокий уровень самоорганизации, что сопровождается эволюцией механизма базисной реакции за счет усложнения состава и структуры катализаторов. Катализатор может изменяться и под влиянием внешних условий, в частности, в результате изменения химического состава окружающей среды, что обеспечивает поступление в реакционную смесь новых, не участвующих в реакции, но взаимодействующих с катализатором веществ. Если такое изменение случайно приводит к ускорению реакции, то последняя пойдет именно по этому пути, исключая все остальные. А.П. Руденко сформулировал основной закон химической эволюции, согласно которому с наибольшей скоростью и вероятностью образуются те пути эволюционных изменений, на которых происходит максимальное увеличение его активности. Саморазвитие, самоорганизация и самоусложнение каталитических систем происходят за счет постоянного потока трансформируемой энергии, главным источником которого является базисная реакция. Базисная реакция является, таким образом, не только источником энергии, необхо-димой для синтеза новых химических соединений, но и механизмом отбора наиболее совершенных эволюционных изменений в катализаторе. В качестве примера рассмотрим реакцию разложения пероксида водорода
В отсутствие посторонних веществ или ионов Н2О2 – стабильное вещество. Энергия активации реакции его разложения равна 750 кДж/моль. Однако многие вещества и ионы катализируют эту реакцию. В частности, катализаторами разложения пероксида водорода являются соединения железа. При этом относительная каталитическая активность аквакомплекса равна 1, гема – 103 и каталаза – 107. Химическая эволюция привела к появлению биологической формы движения. Это произошло в результате развития химических систем и процессов в них происходящих, а не только веществ. Реализация этого направления химической эволюции началась на самых ранних этапах геологической эволюции Земли в «первичном бульоне». Так назвал А.И. Опарин состав океана, где предположительно, по его гипотезе, зародилась жизнь. Химический состав этого водного раствора вначале определялся составом атмосферы, которая образовалась вследствие дегазации недр мантии вулканического происхождения. Атмосфера первичной Земли состояла из паров воды, СО2 и других газообразных соединений (СО, Н2, N2, Н2S, СН4, NH3, HF, HCl, Ar). Вода Мирового океана стала активно взаимодействовать с горными породами, вследствие чего образовались соли Ca, Мg, Fе (реакции нейтрализации) и водорастворимые соли щелочных металлов. Вопросы, связанные с предбиологической эволюцией органической материи, будут рассмотрены в следующем разделе. Возникновение открытых каталитических систем и их отбор по наиболее перспективным для химической эволюции базисным реакциям показали эксперименты С. Миллера и С. Фокса. Воздействие излучения, электрических разрядов и токов высокой частоты на смесь газов первичной атмосферы приводит к образованию различных органических соединений. На первом этапе из воды, оксидов углерода, метана, водорода и аммиака образуются циановодород, муравьиная кислота, формальдегид, уксусная кислота и др. Затем из них были получены глицин, аланин, аспарагиновая кислота, глутаминовая кислота, нуклеотиды и т.д. Образование многих метаболитов, общих для всех живых организмов, в условиях «первичного бульона» можно представить следующим образом. В некоторых районах подводных вулканов могли возникнуть условия спонтанного и длительного протекания какой-либо химической реакции, обеспечиваемой постоянным притоком реагирующих веществ и наличием простейшего катализатора. Простейшими по составу катализаторами базисных реакций с учетом водной среды были, очевидно, гидратированные ионы, в частности, аквакомплексы ионов переходных металлов. Их каталитическая активность могла увеличиваться при замене воды во внутренней сфере комплекса на другие неорганические вещества. Особенно широкие возможности представила замена неорганических лигандов (окружение центрального иона) на органические. Это связано не только с большим разнообразием органических веществ, присутствующих во внешней среде, но и с большими возможностями взаимных превращений органических веществ. Особое значение имели такие изменения катализатора – состава и строения его компонентов, – которые требуют затрат энергии. Эти затраты компенсируются возрастанием скорости базовой реакции, что, в свою очередь, увеличивает возможности образования именно данного катализатора и дальнейшее ускорение базовой реакции. Возникает положительная обратная связь базовая реакция – катализатор. Такая система эволюционирует в направлении усложнения катализатора и повышения его активности. Известно, что при возможности протекания параллельных реакций с разными катализаторами максимальное количество вещества реагирует на пути с наибольшей скоростью. Происходит, таким образом, отбор катализаторов на максимальную скорость базовой реакции. Благодаря сопряжению с базисной реакцией (за счет ее энергии), могут произойти требующие энергии реакции дегидратационной конденсации аминокислот с образованием полипептидов, моносахаров с образованием полисахаридов, органических азотистых оснований, сахаров и фосфорной кислоты с образованием нуклиновых кислот и полинуклеотидов и т.д. Однако увеличение скорости базовой реакции не может быть беспредельным. На некотором этапе увеличение каталитической активности окажется бесполезным, так как лимитирующей стадией процесса будет ограниченность ресурсов питания системы компонентами базовой реакции, скорости их подвода к месту осуществления реакций. Единственным эволюционным изменением такой системы, которое могло бы привести к увеличению ее производительности, служит появление пространственной редупликации сложной системы в целом, т.е. появление функции самовоспроизведения сложных каталитических систем, их размножение.
|
||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2021-05-12; просмотров: 72; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.15.143.181 (0.085 с.) |
 рассматриваемой системы, знака этого изменения, а также константы равновесия. Как было рассмотрено ранее, химическая реакция может происходить, если она приводит к уменьшению свободной энергии (
рассматриваемой системы, знака этого изменения, а также константы равновесия. Как было рассмотрено ранее, химическая реакция может происходить, если она приводит к уменьшению свободной энергии ( ).
).  , то изменение системы не происходит и система находится в равновесии.
, то изменение системы не происходит и система находится в равновесии.
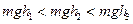 ,
, высоты центра тяжести при опрокидывании тел 1, 2, 3 соответственно.
высоты центра тяжести при опрокидывании тел 1, 2, 3 соответственно. , совершив работу
, совершив работу 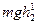 . При опрокидывании тела его центр тяжести описывает некоторую траекторию, и тело начинает падать само, как только центр тяжести пройдет высшую точку траектории. При этом освобождается потенциальная энергия
. При опрокидывании тела его центр тяжести описывает некоторую траекторию, и тело начинает падать само, как только центр тяжести пройдет высшую точку траектории. При этом освобождается потенциальная энергия  . Поскольку вначале была уже затрачена энергия
. Поскольку вначале была уже затрачена энергия  , т.е. в теплоту превращается только эта энергия. Еще более устойчиво тело 3 из-за наличия подставки, поскольку для его опрокидывания необходимо затратить большую энергию
, т.е. в теплоту превращается только эта энергия. Еще более устойчиво тело 3 из-за наличия подставки, поскольку для его опрокидывания необходимо затратить большую энергию  .
. или
или 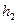 или
или  , и только после этого начнет выделяться энергия, обусловленная разностью энергии начального и конечного состояний. Из этого примера видно, что стабильность состояния зависит не столько от потенциальной энергии тела, соответствующей этому состоянию, сколько от величины энергии активации. Этот пример помогает понять, почему не происходит самопроизвольных превращений во всех веществах, которые способны к таким превращениям с точки зрения запасенной (потенциальной) химической энергии. Хотя в результате превращения энергетически нестабильных соединений в стабильные и выделяется энергия, но для начала этого процесса вещество необходимо активировать, затратив на это определенную энергию активации, которая, однако, не учитывается в суммарном энергетическом балансе процесса.
, и только после этого начнет выделяться энергия, обусловленная разностью энергии начального и конечного состояний. Из этого примера видно, что стабильность состояния зависит не столько от потенциальной энергии тела, соответствующей этому состоянию, сколько от величины энергии активации. Этот пример помогает понять, почему не происходит самопроизвольных превращений во всех веществах, которые способны к таким превращениям с точки зрения запасенной (потенциальной) химической энергии. Хотя в результате превращения энергетически нестабильных соединений в стабильные и выделяется энергия, но для начала этого процесса вещество необходимо активировать, затратив на это определенную энергию активации, которая, однако, не учитывается в суммарном энергетическом балансе процесса.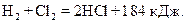
 (93)
(93)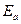 , необходимо проинтегрировать уравнение (93) от
, необходимо проинтегрировать уравнение (93) от  . Интегрирование дает долю молекул в системе с энергиями, большими энергии активации
. Интегрирование дает долю молекул в системе с энергиями, большими энергии активации  . Эти молекулы можно назвать «активными», поскольку энергия, выделяемая при их столкновениях, оказывается достаточной для разрыва или ослабления химических связей.
. Эти молекулы можно назвать «активными», поскольку энергия, выделяемая при их столкновениях, оказывается достаточной для разрыва или ослабления химических связей. Рисунок 118
Рисунок 118
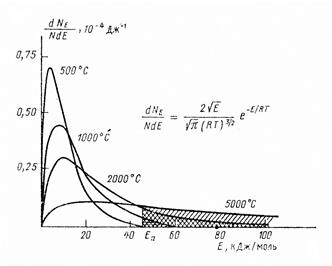 Рисунок 119
Рисунок 119
 .
.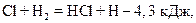
 ,
, – для гомогенных реакций. Реакции гомогенные, если все ре-агенты находятся в одной фазе (жидкой, газообразной или твердой).
– для гомогенных реакций. Реакции гомогенные, если все ре-агенты находятся в одной фазе (жидкой, газообразной или твердой).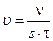
 .
. ,
, ; если С – концентрация одного из продуктов реакции, то
; если С – концентрация одного из продуктов реакции, то  . При таком определении скорость реакции зависит от выбора вещества, изменение концентрации которого рассматривается. Например, в реакции
. При таком определении скорость реакции зависит от выбора вещества, изменение концентрации которого рассматривается. Например, в реакции 

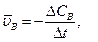
 видно, что
видно, что  равна
равна  и в 2 раза меньше, чем
и в 2 раза меньше, чем  .
.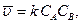

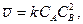
 и
и  ; в реакции
; в реакции  и т.д.
и т.д.
 – константа;
– константа;  – энергия активации.
– энергия активации.

 и
и  – константы скорости некоторой реакции при температурах
– константы скорости некоторой реакции при температурах  и
и 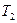 соответственно;
соответственно; 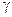 – коэффициент Вант-Гоффа.
– коэффициент Вант-Гоффа.
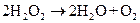 ) в чистом водном растворе составляет 75 кДж/моль, в присутствии коллоидной платины – 49 кДж/моль, а в присутствии фермента каталазы – всего только 23 кДж/моль.
) в чистом водном растворе составляет 75 кДж/моль, в присутствии коллоидной платины – 49 кДж/моль, а в присутствии фермента каталазы – всего только 23 кДж/моль. ), которые взаимодействуют с кислородом с образованием воды.
), которые взаимодействуют с кислородом с образованием воды. ,
,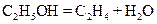 ,
, .
.



