Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Наследственные генные болезни
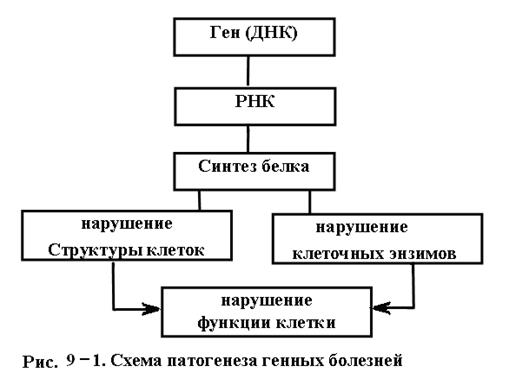
Генные болезни - это те заболевания, которые вызываются генными мутациями. Последние передаются из поколения в поколение без изменений. Существует более 2000 разнообразных наследственных заболеваний человека, характеризующихся различными нарушениями обмена веществ, системы крови, органов чувств, нервной и других систем. Общая частота генных болезней в популяциях равна примерно 1-2 %, в то время как отдельные формы наследственной патологии встречаются значительно (в десятки – сотни - тысячи раз) реже. Возникшие под влиянием мутагенов в гене мутации обычно приводят как к количественным, так и качественным нарушениям в синтезируемом ферменте, белковом продукте. Это обязательно сказывается в виде того или иного нарушения структуры, метаболизма и функций, соответствующего той или иной картине наследственной патологии (рис.9-1). Таким образом в патогенезе генных болезней особое место занимают, во-первых, наследственные ферментопатии (энзимопатии) - наследственные заболевания, обусловленные отсутствием какого-либо фермента или существенным изменением его активности, во-вторых, те или иные структурные нарушения клеток. Развитие патологических, как и нормальных наследственных признаков можно выразить общей схемой: ген ® фермент ® биохимическая реакция ® признак. Наследственные болезни клинически могут обнаруживаться в различном возрасте, что зависит не только от степени, локализации и характера изменения наследственного аппарата, но и от условий жизни (питания, работы, отдыха, состояния окружающей среды, вида и характера повреждений и др.). В зависимости от количества генных мутаций выделяют моногенные и полигенные болезни.
Моногенные являются истинно наследственными заболеваниями (с полностью сформированным дефектом метаболизма, структуры и функции), передающимися в ряду поколений. Полигенные чаще относятся к болезням с наследственным предрасположением (с незначительным дефектом метаболизма, структуры и функции), причем эта предрасположенность обычно является многофакторной. Принципиально каждый из имеющихся у человека около 70000 генов может мутировать, а значит приводить к появлению нового или исчезновению имеющегося белка. В связи с этим можно полагать, что количество наследственных болезней, вызванных генными мутациями, может быть значительно больше выявленных к настоящему времени.
Для многих генных болезней идентифицирован первичный аномальный продукт гена или ведущее патогенетическое звено на биохимическом уровне. Последние классифицируют в зависимости от вида пораженных (измененных) белков: структурных, транспортных, ферментных. Например, при синдроме Элерса-Данлоса изменяется молекулярная структура коллагена. Это приводит к повышенной эластичности кожи, подвижности суставов, растяжимостью хорд сердечных клапанов, а также подвывихом хрусталика, отслойкой сетчатки. Поражение транспортных белков (диаминокислот: лизина, аргинина, орнитина) отмечается при лизинурической непереносимости белка. Наиболее обширную и хорошо изученную группу моногенных заболеваний составляют энзимопатии. Исходя из гипотезы «один ген - один фермент» уже расшифрованы многие дефекты ферментов, обусловившие нарушения обмена углеводов (гликогена), липидов, белков, в том числе гликопротеидов, аминокислот, гормонов и др. Следует, однако, указать, что у многих моногенных наследственных болезней до сих пор не выявлены первичные биохимические дефекты (например, при ахондроплазии - наследственной болезни костной системы, проявляющейся низким ростом, аномальным развитием хрящевой ткани, особенно в эпифизах трубчатых костей. При нормальной длине туловища больные имеют укороченные, деформированные и бугристые конечности). Как аномалии, так и болезни могут наследоваться по аутосомно-доминантному типу, аутосомно-рецессивному типу, а также сцеплено с полом (т.е. передаваться с половой, главным образом, с Х- хромосомой). 9.3.1. Виды и пути передачи наследственной патологии По аутосомно-доминантному типу ( рис.9-2) наследуются обычно не опасные для жизни изменения, аномалии, а также болезни и синдромы, представляющие различную степень опасности для организма. К аномалиям относятся: короткопалость, многопалость, сросшиеся пальцы, искривление ногтей, костей, ушных раковин, близорукость, дальнозоркость, астигматизм и др.
К болезням относятся: врожденные катаракта, глаукома, отосклероз, мышечная атрофия, мышечная дистрофия, полипоз толстой кишки, серповидноклеточная анемия (HbS), муковисцедоз, талассемия, хондродистрофии, ахондроплазии, ретинобластома и др. К синдромам относятся: § синдром Марфана (подвывих хрусталика, паучьи пальцы, аневризма аорты, возникающие из-за нарушения синтеза белков в соединительно-тканных структурах), § синдром Гольденара (расщепление губы и неба, множественные базально-клеточные карциномы, кисты челюсти, аномалии скелета), § синдром Горлина (расщепление губы и неба, односторонняя дисплазия ушной раковины, аномалии позвоночника, сердца, почек и гениталий), § синдром акроостеолиза (расщепление неба, «растворение» концевых фаланг с утолщением пальцев, низкий рост, преждевременное выпадение зубов, долихоцефалия), § синдром ключично-черепной дисплазии (расщепление неба, широкий свод черепа (незаращенные роднички на черепе, маленькое лицо, отсутствие ключиц) и др. Эти аномалии, заболевания и симптомы могут передаваться по типу как полного, так и неполного доминирования. Степень проявления доминантного признака в фенотипе может быть различной (незначительной или сильной). Последнее определяется не только генетическими факторами, но и факторами внешней среды.
Рис. 9-2. Аутосомно-доминантный тип наследования
По аутосомно-рецессивному типу (рис.9-3) передается большинство наследственных болезней, которые развиваются у гомозиготных детей, оба родителя которых являются гетерозиготными носителями патологического признака и фенотипически здоровы. Проявление патологического гена характеризуется пенетрантностью (вероятностью фенотипического проявления гена - отношением числа больных особей к числу носителей генов) и экспрессивностью (степенью развития признака, контролируемого данным геном). Патологический ген чаще всего встречается у детей, имеющих кровное родство родителей, обладающих одинаковым рецессивным патологическим признаком.
Рис. 9-3. Аутосомно-рецессивный тип наследования
По аутосомно-рецессивному типу передается аномалия в виде альбинизма (отсутствие пигмента в коже, волосах, радужке глаза из-за отсутствия тирозиназы, в норме превращающей тирозин в меланин). По данному типу передается много наследственных аутосомно-рецессивных заболеваний, таких как: врожденная глухонемота, идиотия со слепотой, шизофрения, сахарный диабет, полная цветовая слепота, микроцефалия и др.
Очень часто по аутосомно-рецессивному типу передаются различные нарушения обмена веществ: § фенилкетонурия (основу которой составляет понижение активности глюкозоаланингидроксилазы, что приводит к накоплению l-фенилаланина в тканях из-за блокады его перехода в тирозин), § генерализованный гликогеноз (понижение активности глюкозо-6-фосфатазы органов, из-за чего гликоген накапливается в тканях), § галактоземия (возникает из-за дефекта лактазы - фермента, расщепляющего лактозу; характеризуется также увеличением печени, развитием катаракты и психических отклонений), § сфинголипидоз (возникает из-за отсутствия фермента сфинголипазы в клеточных мембранах, способствует отложению холестерина и нарушению обмена липидов как мембранных сосудов, так и других клеточных структур; сопровождается гибелью детей в возрасте до 5 лет, § дефицит пиридоксина - витамина В6 (приводит к нарушению обмена белков, аминокислот, липидов, ферментов, развитию гипохромной анемии, эпитептиформных судорог и др.) § адреногенитальный синдром: генетически обусловленная блокада синтеза глюкокортикоидных гормонов в коре надпочечников (возникает в результате дефицита А-В-гидроксилазы), сопровождающаяся увеличением в последней продукции андрогенов. Это приводит к маскулинизации девочек и преждевременному половому созреванию мальчиков. Наследование болезни, сцепленное с полом, связанно, главным образом, с половой Х-хромосомой (рис.9-4). Большинство наследственных болезней (тех или иных патологических признаков), связанных с полом, передаются рецессивно. Таких болезней насчитывается около 100. Женщина-носительница патологического признака сама не страдает, так как здоровая Х-хромосома доминирует и подавляет Х-хромосому с патологическим признаком, т.е. компенсирует неполноценность данной хромосомы. При этом болезнь проявляется у лиц мужского пола. По рецессивному сцепленному с Х- хромосомой типу, передаются: дальтонизм (красно-зеленая слепота), атрофия зрительных нервов, куриная слепота, миопия Дюшена, синдром «курчавых волос» ( возникает в результате нарушенияобмена меди, повышения ее содержания в тканях. Проявляется слабоокрашенными, редкими и выпадающими волосами, умственной отсталостью и т.д.), дефект ферментов переводящих пуриновые основания в нуклеотиды (сопровождается нарушением синтеза ДНК в виде синдрома Леша-
Рис. 9-4. Наследование, сцепленное с полом
Кайана, проявляющегося умственной отсталостью, агрессивным поведением, членовредительством), гемофилия А (в результате недостатка антигемофильного глобулина - фактора. VIII), гемофилия В (в результате дефицита фактора Кристмаса - фактора IХ) и т.д. По доминантному сцепленному с Х-хромосомой типу передаются гипофосфатемический рахит (не поддающийся лечению витамином D2 и D3), коричневая эмаль зубов и др. Данное заболевание развивается у лиц и мужского, и женского пола. Болезни с наследственным предрасположением возникают у лиц, имеющих незначительную неполноценность той или иной наследственной структуры, которая в условиях нормальной жизнедеятельности клинически не проявляется (т.к. способна компенсироваться). Однако под влиянием различных неблагоприятных внешних воздействий (тех или иных значительных нагрузок) эта наследственная неполноценность реализуется в виде определенного полома метаболических процессов, структуры и функции, способного привести к развитию соответствующего заболевания. Значимую роль в наследственной предрасположенности обычно играют измененные конституция, реактивность организма, а также различные отрицательные влияния внешней среды и др. Эти заболевания представляют довольно обширную группу (по данным ВОЗ составляют более 90 %) наследственной патологии, отличающихся многообразием своих проявлений. Заболевания с наследственным предрасположением могут быть моногенными, но чаще полигенными (т.е. вызываться мутацией соответственно одного, либо многих генов) и вызываться разными патогенными для организма факторами (грязные воздух, вода, пища; непереносимость молока, молочных продуктов, лекарств и т.д.). Неслучайно эти заболевания называются мультифакторными. Например, часто встречаемая в разных странах мира непереносимость различными людьми молока, молочных продуктов и молочной пищи обычно обусловлена аутосомно-рецессивным признаком непереносимости глюкозы из-за отсутствия или угнетения b-галактозидазы в кишечнике гомозитотных организмов. К болезням с наследственной предрасположенностью относятся: сахарный диабет, язвенная болезнь желудка и двенадцатиперстной кишки, гипертоническая болезнь, атеросклероз, подагра, туберкулез, бронхиальная астма, шизофрения, псориаз, коллагенозы и другие формы патологии . Хромосомные абберации Хромосомные аберрации (от лат. aberratio - отклонение) или хромосомные болезни и синдромы -это болезни и синдромы, обусловленные индивидуальным отклонением от нормы структуры, количества и функции хромосом. Эти нарушения хромосом возникают при нарушении созревания и деления половых клеток (гамет) родителей (в процессе их мейоза) или на стадиях дробления зиготы (оплодотворенной яйцеклетки). Данные перестройки хромосом, как правило, дают жизнеспособную половую клетку. Известно, что около 17 % эмбрионов и плодов погибают до рождения, из них около 40 % в результате хромосомных нарушений. Число хромосомных болезней превышает 500.
Показано также, что подавляющая часть хромосомных аномалий относятся к категории летальных мутаций. В этой связи, для характеристики их количественных параметров используются два показателя - частота распространения и частота возникновения. Однако, если половая клетка оказывается жизнеспособной и оплодотворенной, то все равно эмбрион, либо плод чаще погибают внутриутробно. Даже когда организм остается живым, у него всегда сильно нарушается и соматическое, и психическое развитие. Редко (до 3-5 % случаев) такие организмы сами способны вырастить потомство, которое, однако, может наследовать эту же патологию. Часть хромосомных аберраций может клинически не проявляться. В основе хромосомных болезней и синдромов лежат нарушения либо числа хромосомных наборов (в виде тетраплоидии и триплоидии), либо числа отдельных хромосом (трисомия - наличие добавочной хромосомы в диплоидном наборе или моносомия - одна из хромосом отсутствует), либо изменения (в сторону, как увеличения, так и уменьшения) части той или иной хромосомы. Структурные перестройки хромосом составляют самую многочисленную группу хромосомных болезней. Большинство хромосомных болезней не передаются в ряду поколений. Нарушения хромосомных комплексов (гетероплоидия) в виде нерасхождения или неправильного расхождения хромосом может коснуться любой пары в хромосомном наборе - как соматических (аутосомных), так и половых (рис.9-5). Нерасхождение больших соматических хромосом (групп А, В, С) всегда дает нежизнеспособную либо половую клетку, либо зиготу, либо приводит к гибели внутриутробно развивающегося организма. При нерасхождении малых хромосом (групп D, Е, F, G) половая клетка часто жизнеспособна и может даже дать жизнеспособное потомство. Однако излишек или дефицит хромосом, как правило, проявляется той или иной хромосомной болезнью или синдромом.
Гетероплодия по аутосомам
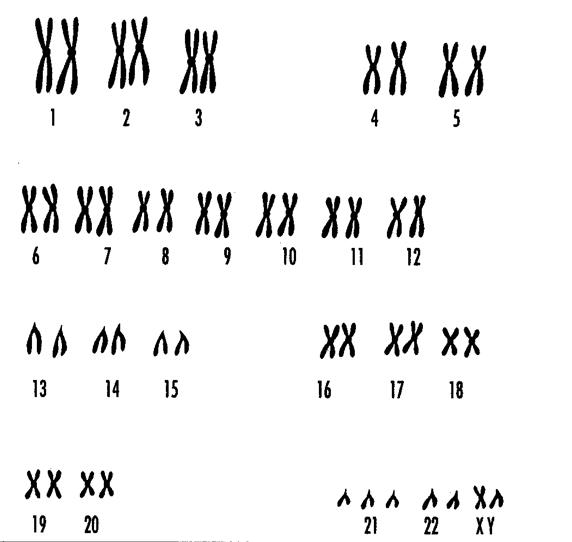
Может проявляться либо в виде полисомий, либо в виде моносомий. Среди гетероплоидий в виде полисомий по аутосомам наиболее часто встречается синдром Дауна (рис.9-6). Такие больные имеют в кариотипе 47 хромосом, в том числе лишнюю 21-ю аутосомную хромосому, т.е. трисомию по 21-й хромосоме. Больные характеризуются малым черепом, близко расположенными, чаще косыми, глазами с монголоидным разрезом и нависающей складкой кожи над верхним веком, маленьким носом с широкой плоской переносицей, деформированными округлыми, небольшими ушными раковинами, толстыми губами, полуоткрытым ртом с выступающим большим изрезанным языком, низким ростом, короткими конечностями, ладонями, стопами и пальцами (мизинец мал и обычно загнут вовнутрь). Больные отличаются замедленным физическим развитием, нарушениями моторной деятельности, мышечной слабостью. Синдром Дауна считается наиболее распространенной формой умственной отсталости (чаще дебилы, реже имбецилы и идиоты). Обычно имеет место недоразвитие гениталий, дегенерация семенников, задержка полового развития. Часто отмечаются пороки развития сердца, органов пищеварительного тракта и др. Мужчины с синдромом Дауна бесплодны, женщины редко могут иметь потомство.
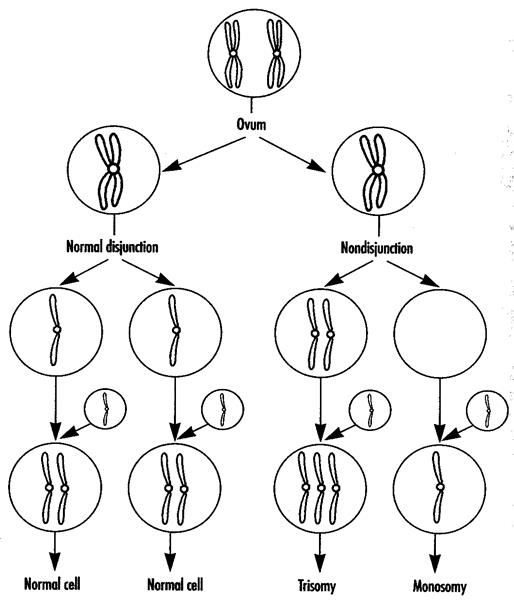
Рис. 9-5. Схема формирования количественных нарушений хромосом
Заболевание встречается с частотой 1:500 – 1:700 новорожденных. Чаще встречается у детей, возраст матери которых превышает 35 лет. Больные с синдромом Дауна обычно поддаются обучению бытовым навыкам, координации движений, речи и другим простым функциям. Реже (с частотой 1:3500 – 1:4000 новорожденных) встречается синдром Патау. Такие больные имеют трисомию по 13-й аутосомной хромосоме. Новорожденные отличаются уменьшенной массой тела, микроцефалией, деформацией мозгового и лицевого черепа, расщеплением верхней губы или неба, различными нарушениями строения глаз, недоразвитыми пальцами. Часто имеют место пороки сердца, легких, почек, матки. Новорожденные обычно погибают в течение нескольких дней или недель. Редко живут несколько лет. На протяжении своей короткой жизни являются умственно отсталыми.
Рис. 9-6. Кариотип больного синдромом Дауна
В эритроцитах превалирует HbF. Гетероплоидия в виде моносомий чаще касается 21-й, 13-й и 18-й аутосомных хромосом. В частности, моносомия по 21-й хромосоме (больные антимонголизмом) характеризуется наличием у больных раскосых глаз, больших оттопыренных ушных раковин, большого носа с широкой переносицей, повышенного мышечного тонуса, выступающего затылка. Больные отличаются низким ростом, умственной отсталостью и частым развитием пороков сердца.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-09-19; просмотров: 486; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.216.239.46 (0.033 с.) |