Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
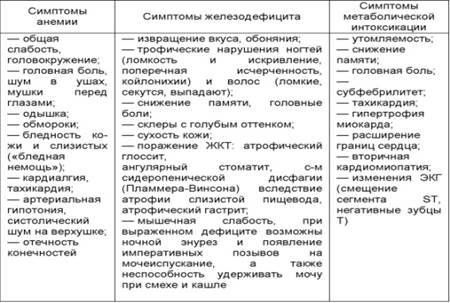
Симптомы железодефицитной анемии
Лабораторные данные. 1. В крови - гипохромная, гипорегенераторная микроцитарная анемия, анизоцитоз, пойкилоцитоз, преобладание микроцитов, возможно ускорение СОЭ. 2. Снижение содержания ферритина (N 15-150 мкг/л) и снижение уровня железа сыворотки крови (в норме 13-30 мкмоль/л). Для получения достоверных результатов в течение 5 дней перед исследованием не принимать препараты, содержащие железо. 3. Железосвязывающая способность сыворотки (общая и латентная) может быть повышена. В норме ОЖСС 30-85 мкмоль/л, латентная ЖСС выше 47 мкмоль/л. Лечение. Существует 5 правил: 1. Не следует лечить только диетой (так как в суточном рационе пищи железа недостаточно для восстановления потери и всасываемость железа из пищевых продуктов ограничена). Без препаратов железа вылечить ЖДА невозможно, даже при употреблении пищи, богатой железом! Следует ограничить прием молочных продуктов за 2 часа до приема препаратов железа. 2. Не следует лечить переливанием эритромассы. Трансфузии делают только при уровне гемоглобина ниже 50 г/л (угроза гипоксической комы) либо перед экстренной операцией. 3. Не следует начинать лечение с парентерального введения железа. Парентеральная ферротерапия показана только при тяжелой непереносимости всех препаратов железа или появлении выраженной диспепсии после приема препаратов. В/в введение препаратов железа может сопровождаться тяжелыми аллергическими реакциями, саркомами мягких тканей, флебитов, абсцессов в местах в/м введения железосодержащих препаратов. 4. Основная патогенетическая терапия состоит в пероральном приеме железосодержащих препаратов. При выборе суточной дозировки следует ориентироваться на общее содержание в нем железа и на количество двухвалентного железа, содержащегося в выбранном препарате железа (какие, см. ниже). Суточная доза элементарного (двухвалентного) железа 100-300 мг. Препарат железа (ПЖ) должен содержать оптимальное количество двухвалентного железа; хорошо переноситься больными, обладать минимальными побочными эффектами; быть удобным для приема; содержать дополнительные компоненты, улучшающие всасывание и биодоступность железа; при этом не содержать компоненты, затрудняющие всасывание железа или конкурирующие с ним.
5. Основной (насыщающий) курс ферротерапии обычно составляет 1,5-2 мес. Контроль эффективности лечения проводят по приросту гемоглобина на 21 день лечения. После нормализации уровня гемоглобина необходимо снизить суточную дозу препарата в 2 раза и продолжить поддерживающее лечение в течение 2-4 мес. Лечение начинают с приема препаратов железа в максимально переносимой дозе до полной нормализации содержания гемоглобина. Не следует принимать одновременно пищевые продукты и медикаменты, уменьшающие всасывание железа (крепкий чай и кофе, молочные продукты, фосфорная кислота, тетрациклины, альмагель, циметидин, соли кальция, магния, алюминия). Для лечения железодефицитной анемии используют ПЖ: монокомпонентные, содержащие одно железо (Ферроградумет, Ферронат, Хеферол), сложные - с включением в состав аскорбиновой или янтарной к-т, фруктозы или др. компонентов, усиливающих всасывание железа (Сорбифер Дурулес, Ферроплекс), и комбинированные, содержащие также витамины В12, В6, В1, фолиевую кислоту, лизин, микроэлементы (Фенюльс, Фенотек, Актиферрин, Ранферон-12). Основные препараты железа представлены в виде сульфата, глюконата, хлорида, фумарата железа. Любой препарат лучше рекомендовать за 0,5-1 час до еды или не ранее 2 часов после еды. Суточная доза железа определяется из расчета 2-3 мг/кг для взрослых. Для детей доза несколько выше: до 3-х лет — 5-8 мг/кг, 4-7 лет — 4-6 мг/кг, 8-16 лет — 3-5 мг/кг. При создании препаратов железа для лучшей переносимости и профилактики перегрузки организма железом стояла задача: снизить дозу железа без ущерба для эффективности. С этой целью, например, комбинируют железо с аминокислотами (D-, L-серин). В препарат Тотема, помимо железа, входит также медь и марганец (все в виде органических соединений — глюконатов). При назначении ПЖ в достаточной дозе в ходе лечения контролируется показатель ретикулоцитов. Их число на 7-10 день лечения повышается (ретикулоцитарный криз) и возрастает к 21 дню. Уровень Hb повышается на 3-4 неделе лечения, у некоторых больных на 6-8 неделе. После основного курса ферротерапии - поддерживающий курс в течение 1,5-3 мес, доза при этом уменьшается в 2 раза.
Профилактику железодефицитной анемии проводить у след. групп: доноры, беременные и кормящие грудью, девочки-подростки с обильными менструациями, женщины с обильными и длительными менструациями, с фибромиомой матки. Первичная профилактика заключается в рациональном питании. Всем лицам, имеющим факторы риска (после операций на ЖКТ, при полименорреи, во время беременности, лактации) проводится первичная профилактика препаратами железа. По рекомендациям ВОЗ все беременные на протяжении II—III триместров и в первые 6 мес лактации должны получать ПЖ. Вторичная профилактика железодефицитной анемии заключается в проведении поддерживающей ферротерапии после нормализации Hb еще 1-3 мес, противорецидивной терапии 1 раз в 3-6 мес, в профилактическом приеме при полименоррее препаратов железа женщинами после каждых менструаций в дозе 30-60 мг элементарного железа 7-10 дней. При рецидивирующих кровотечениях, например, геморроидальных, курс лечения железом в течение 6 мес и более. 39. В12-дефицитые и фолиево-дефицитные анемии: классификация, этиология, патогенез, клиника, диагностика, терапевтическая тактика (терапия насыщения и поддержания). Мегалобластные анемии - группа заболеваний, хар-ся появлением в красном костном мозге мегалобластов - клеток красного ядра больших размеров с измененной структурой ядра, которые прослеживаются на всех стадиях дифференцировки эритроидных предшественников. Появление мегалобластов связано с нарушением синтеза ДНК и замедлением созревания клеток. Витамин B12-дефицитная анемия - группа заболеваний, связанных с дефицитом цианокобаламина (витамин B12) или нарушением его метаболизма. Причины дефицита витамина В12: 1) неполноценное питание (исключительно вегетарианское); Клиника. Болеют лица в возрасте 45-65 лет, редко клиника встречается у людей до 30 лет, чаще болеют женщины. Обычно манифестация (прогрессия) наступает весной или осенью. Заболевание развивается постепенно, нередко долгое время болезненное состояние проявляется поражением ЖКТ, иногда признаками поражения нервной системы (радикулиты), адаптированной (легкой степени) анемией. Выражены синдромы: поражение кроветворной системы, ЖКТ и нервной системы. Клиника проявляется слабостью, одышкой, тахикардией, периодической диареей, жгучими болями в языке при приеме острой и кислой пищи, парестезиями, снижением чувствительности, а при тяжелых анемиях - спутанностью сознания, депрессией, деменцией. Объективно: покровы бледные с матовым и лимонно-желтым оттенком. Язык гладкий, сглаженный, атрофичный (иногда, гиперпластичный), блестящий, влажный, иногда красный и воспаленный («лакированный» язык, глоссит Хантера). Селезенка часто увеличена, иногда увеличена печень. Парестезии пальцев рук и ног. Атрофия мышц, полиневриты, расстройства координации (неуверенная или шаткая походка, нескоординированные движения).
Диагностика: -очень низкое содержание витамина В12 в плазме (менее 100 пг/л, N 160-950 пг/л), увеличение ферритина, уменьшение гаптоглобина, повышение ЛДГ; - антитела к внутреннему фактору или к париетальным клеткам в сыворотке крови (обнаруживаются в 50% случаев). - гиперхромия, макроцитарная гипо-, норморегенераторная (диспластическая) анемия. Обнаруживаются мегалоциты (11-14 мкм), макроформы и анизоцитоз, включения в эритроцитах (тельца Жолли, кольца Кебота, зернистость Гейнца), полисегментация нейтрофилов. - мегалобластный костный мозг («синий» при окраске по Романовскому); - отрицательный тест Шиллинга (при экскреции мочи после приема радиоактивного витамина В12 внутрь уменьшается выделение кобаламина); - при Ph-метрии желудочного сока гипо- и ахлоргидрия; - при биопсии слизистой желудка - фундальный гастрит, гипертрофия бокаловидных клеток, атрофия париетальных и главных клеток, клеточный атипизм; - увеличение билирубина за счет непрямого (неконъюгированного). Лечение. Режим амбулаторный или стационарный в зависимости от тяжести анемии. Диета: с повышенным содержанием белков, преимущественно продукты животного происхождения. Лекарственная терапия заключается в назначении цианкобаламина по 1000 мкг в/м 1 раз в день, далее по 500 мкг 3-4 недели, затем по 200 мкг/сут в течение 1-1,5 месяца или по 500 мкг 1 раз в неделю 1 месяц. С целью профилактики рецидивов пожизненно вводят цианкобаламин по 500 мкг 2 раза в месяц. Уже в первые сутки после в/м введения витамина В12 происходит трансформация мегалобластного типа в нормоцитарный, усиливается эритропоэз. Через 48-72 часа после первой инъекции витамина В12 начинает увеличиваться число ретикулоцитов. Ретикулоцитарный криз нарастает к 7-12 дню, что может быть оценено как результат эффективной терапии. В клинических условиях фолиевая кислота не должна использоваться для лечения В12-дефицитной анемии, так как могут развиться серьезные неврологические расстройства даже при ликвидации анемии. Трансфузии эритроцитов проводят при угрозе анемической комы — наиболее грозного и плохо поддающегося лечению осложнения В12-дефицитной анемии. Прогноз при адекватном лечении благоприятный. Хронизация анемии отмечается при алкоголизме и у лиц после тотальной резекции желудка или кишечника. После достижения ремиссии больные подлежат диспансерному наблюдению, проведению ФГС с биопсией слизистой желудка и противорецидивному лечению витамином В12 весной и осенью.
Фолиеводефицитная анемия - анемия, обусловленная дефицитом фолиевой кислоты или нарушения ее утилизации в процессе эритропоэза, что приводит к мегалобластному типу кроветворения. Патогенез. Фолиевая кислота и ее соединения известны под названием фолатов. Организм получает фолаты при расщеплении содержащихся в пище полиглютаматов в моноглютаматы в тонком кишечнике. В плазме происходит превращение метилтетрафолата в присутствии витамина В12 в тетрагидрофолаты. Последние превращаются в формил-ТГФ (фолиновая кислота), затем в дезокситимидин-монофосфат с последующим синтезирующим действием на ДНК. Таким образом, дефицит витамина В12 ведет к нарушению синтеза фолиновой кислоты и развитию В12-дефицитной анемии. Дефицит фолиевой кислоты ведет к развитию фолиеводефицитной анемии. Суточная потребность фолатов составляет 100 мкг. Запасы, создаваемые в тканях (печень), достаточны для синтезирования ДНК в течение 1-3 мес. Этиология. 1. Недостаток фолиевой кислоты в пище (в том числе вскармливание новорожденных козьим или порошковым молоком). 2. Нарушения всасывания в тонком кишечнике и нарушения депонирования в печени (в том числе при злоупотреблении алкоголем). 3. Прием антагонистов фолиевой кислоты (метотрексата), аналогов пурина и пиримидина, противосудорожных препаратов - дифенина, фенобарбитала. 4. Повышение потребности в фолиевой кислоте (беременность, новорожденные, миелопролиферативные синдромы, хронический гемолиз). 5. При очень активной пролиферации клеток (гемолиз, лейкозы и другие опухоли, инфекции, псориаз). Клиника сходна с В12-дефицитной анемией. Не характерен атрофический гастрит с ахилией, нет фуникулярного миелоза. Не наблюдается геморрагический диатез. Более выражены функциональные признаки поражения ЦНС. Встречается чаще у детей, молодых женщин, алкоголиков, больных эпилепсией. Диагностические критерии дефицита фолиевой кислоты: низкий уровень содержания фолатов в сыворотке (натощак) 3-25 нг/мл; низкий уровень содержания фолатов в эритроцитах (N 100-415 нг/мл); макроцитарная диспластическая анемия; мегалобластный костный мозг; нормальный или несколько сниженный уровень витамина В12 в сыворотке крови (менее 100 мг/мл, N 160-930 мг/мл). Гемограмма и миелограмма сходны с витамин В12-дефицитной анемией. Дифференциальный диагноз с В12-дефицитной анемией представлен в таблице. Основные дифференциально-диагностические признаки витамин В12- (2 столбик) и фолиеводефицитной анемий (3 столбик)
Лечение. Фолиевая кислота внутрь 5-15 мг/сут на протяжении 4-6 недель до получения ремиссии. В последующем при неустраненной причине — поддерживающая терапия 1-5 мг/сут.
40. Хронический миелолейкоз: этиология, патогенез, роль хромосомных аббераций в развитии лейкоза, фазы лейкемического процесса, терапия 1-ой линии, терапия резерва. Показания к трансплантации костного мозга, исходы и осложнения ХМЛ. Хронический миелолейкоз (ХМЛ) – хронически протекающее миелопролиферативное заболевание, при котором наблюдается повышенное образование гранулоцитов, преимущественно нейтрофилов, являющихся субстратом опухоли. Источник опухоли – клетка-предшественник миелопоэза. Этиология. Причиной патологического роста клеток считается мутация клетки-предшественника миелопоэза (частично детерминированная полипотентная клетка). Это доказывается обнаружением у больных ХМЛ патологической Ph-хромосомы (филадельфийской) в клетках миелоидного, эритроидного, моноцитарного и тромбоцитарного рядов. Ph-хромосома является частым клеточным маркером, подтверждающим происхождение всего патологического клона клеток при ХМЛ от одной материнской. Несмотря на то, что лейкозными являются все три ростка костного мозга, в развернутой стадии ХМЛ наблюдается безграничный рост, только одного ростка – гранулоцитарного. Существенно повышается продукция и мегакариоцитов (тромбоцитов). Классификация. Заболевание закономерно проходит в своем развитии две стадии – моноклоновую (развернутая доброкачественная) и поликлоновую (терминальную злокачественную). Этому соответствуют три фазы хронического миелолейкоза в клиническом отображении: - хроническая –гиперплазия гранулоцитарного ростка кроветворения в красном костном мозге, при этом способность клеток к дифференцировке и созреванию сохранена; миелоидная пролиферация костного мозга + небольшие изменения в крови без явлений интоксикации; наблюдается умеренный лейкоцитоз, со сдвигом лейкоцитарной формулы до миелоцитов, увеличением содержания зрелых и созревающих гранулоцитов в красном костном мозге, эритро- тромбоцитопоэз сохранены, селезенка нормальных размеров; - фаза акселерации – через 3-3,5 года выраженные клинико-гематологические проявления (интоксикация продуктами распада лейкозных клеток, увеличение печени и селезенки, миелоидная пролиферация костного мозга + изменения в крови: появляются бластные клетки, анемия, тромбоцитопения; нарастают симптомы интоксикации - лихорадка, потливость, слабость, снижение массы тела. Важный признак - резистентность к успешно применявшейся ранее химиотерапии); - бластный криз или терминальная (соответствует развитию поликлоновой опухоли) – рефрактерность к проводимой цитостатической терапии, истощение, значительное увеличение селезенки и печени, дистрофические изменения внутренних органов, выраженные изменения крови (анемия, тромбоцитопения). Появление в периферической крови бластных клеток (до 30–90%), в связи с чем заболевание приобретает черты острого лейкоза. Чаще всего в костном мозге и периферической крови бластный криз хар-ся появлением миелобластов, однако могут встретиться и недифференцируемые бластные клетки. Одновременно происходит значительное угнетение тромбоцитопоэза, развивается геморрагический синдром. Клиническая картина. Миелопролиферативный синдром (обусловлен миелоидной пролиферацией костного мозга) включает: а) общие симптомы, вызванные интоксикацией, разрастаниями лейкозных клеток в костном мозге, селезенке и печени (потливость, слабость, снижение массы тела, тяжесть и боль в области селезенки и печени), оссалгии. б) увеличение печени и селезенки; в) лейкемические инфильтраты в коже; г) характерные изменения в костном мозге и периферической крови. Синдром, обусловленный осложнениями: а) геморрагический диатез (геморрагии и тромбозы вследствие нарушения прокоагулянтного и тромбоцитарного звеньев гемостаза); б) гнойно-воспалительные (пневмонии, плевриты, бронхиты, гнойные поражения кожи и подкожной клетчатки), обусловленные резким снижением активности иммунитета; в) мочекислый диатез (гиперурикемия вследствие повышенного распада гранулоцитов). При исследовании периферической крови обнаруживают: 1) лейкоцитоз (кол-во лейкоцитов колеблется в широких пределах) с появлением в лейкоцитарной формуле пролиферирующих форм (миелобласты и промиелоциты) и созревающих гранулоцитов (миелоциты, метамиелоциты). Функциональные свойства лейкоцитов и содержание в них ферментов изменены: снижена активность щелочной фосфатазы нейтрофилов, нарушена способность к фагоцитозу. 2) имеется базофильно-эозинофильная ассоциация. 3) в ранних стадиях болезни возможно обнаружение гипертромбоцитоза, в дальнейшем - тромбоцитопения. 4) развитие нормоцитарной, нормохромной анемии, связанной в основном с вытеснением лейкозным клоном красного ростка кроветворения, можно наблюдать в развернутой клинико-гематологической стадии. В терминальной стадии анемия становится еще более выраженной. 5) ускорение СОЭ При исследовании костного мозга: 1) обнаруживают миелоидную пролиферацию костного мозга, нормальный миелопоэз полностью замещен патологическим клоном. 2) В мазке костного мозга преобладают гранулоциты: соотношение лейкоциты/эритроциты достигает 10:1, 20:1 за счет увеличения гранулоцитов. 3) Если в периферической крови высокий тромбоцитоз, в костном мозге отмечается большое количество мегакариоцитов. При пункции увеличенной селезенки обнаруживается преобладание миелоидных клеток. Диагностическими критериями заболевания являются: 1) лейкоцитоз более 20 - 103 в 1 мкл крови; 2) появление в лейкоцитарной формуле пролиферирующих форм (миелобласты и промиелоциты) и созревающих гранулоцитов (миелоциты, метамиелоциты); 3) миелоидная пролиферация костного мозга (по данным миелограммы и трепанобиопсии); 4) снижение активности щелочной фосфатазы нейтрофилов (менее 25 ед); 5) обнаружение Ph-хромосомы в кроветворных клетках; 6) расширение плацдарма кроветворения (по данным сцинтиграфии костей); 7) увеличение размеров селезенки и печени. ХМЛ следует дифференцировать от так называемых лейкемоидных реакций, которые могут возникать при ряде заболеваний (туберкулез, рак, различные инфекции, почечная недостаточность и пр.). По определению А. И. Воробьева (1985), лейкемоидная реакция – это «изменения в крови и органах кроветворения, напоминающие лейкозы и другие опухоли кроветворной системы, но не трансформирующиеся в ту опухоль, на которую они похожи». При лейкемоидной реакции наблюдается высокий лейкоцитоз, в периферической крови появляются незрелые нейтрофилы, однако базофильно-эозинофильная ассоциация не обнаруживается. Дифференциальный диагноз основывается на выявлении основного заболевания (рак, туберкулез и пр.), на повышении активности щелочной фосфатазы нейтрофилов (вместо ее снижения при ХМЛ). При стернальной пункции для лейкемоидной реакции характерно увеличение содержания миелоцитов, однако Ph-хромосома никогда не определяется. Лечение. Воздействие на функционирование онкогена препаратом иматиниб мезилат (торговое название - Гливек) - ингибитор ABL-тирозинкиназы. Он соединяется с активными центрами BCR-ABL-тирозинкиназы, что приводит к гибели клеток, содержащих ее, т.е. Ph-положительных клеток. Эффективность превосходит все ранее известные терапевтические средства, применяемые у больных ХМЛ (миелосан, гидроксимочевина, интерферон-α, аллотрансплатация). В настоящее время во всем мире Гливек является препаратом 1-й линии терапии. Назначается по 400 мг в I стадии, по 600 мг во II стадии, до 800 мг в III стадии курсами. Аллотрансплантация гемопоэтических стволовых клеток и препараты новой генерации ингибиторов тирозинкиназ (растительный алкалоид гомогаррингтоин, который проявил высокую эффективность в хронической фазе, в фазе акселерации и даже в бластном кризе ХМЛ. Назначается по 2,5 мг/кг в/в курсом до 14 дней, затем по 7 дней в месяц для поддержания ремиссии) используются в качестве 2-й и последующих линий терапии у больных в хронической фазе ХМЛ с резистентностью к Гливеку или его непереносимостью. ПОКАЗАНИЯ К ТРАНСПЛАНТАЦИИ КОСТНОГО МОЗГА: - Аллогенная трансплантация костного мозга: острые лейкозы; хронический миелолейкоз; тяжелая апластическая анемия; гемоглобинопатии; врожденные иммунодефициты и нарушения метаболизма. - Аутологичная трансплантация костного мозга: злокачественные лимфомы; некоторые солидные опухоли; аутоиммунные заболевания. Прогноз. Длительность жизни больных ХМЛ в среднем составляет 3–5 лет, у отдельных больных достигает 10 лет и более. Осложнения ХМЛ. Острая сердечно-сосудистая недостаточность, инфекционные осложнения, ДВС-синдром и др. Профилактика. Точных мер предупреждения ХМЛ не существует, в связи с чем можно говорить лишь о вторичной профилактике болезни, которая состоит в предупреждении обострений болезни (поддерживающая терапия, исключение инсоляции, простудных заболеваний). 41. Злокачественные неходжкинские лимфомы: классификация, морфологические варианты, клиника, лечение. Исходы. Показания к трансплантации костного мозга. Нелейкемические гемобластозы лимфатического происхождения (неходжкинские лимфомы) разделяют на лимфоцитомы и лимфосаркомы. • Лимфоцитомы - внекостномозговые опухоли, состоящие из зрелых лимфоцитов или лимфоцитов и пролимфоцитов. Это доброкачественные моноклоновые лимфатические опухоли; трансформация в лимфосаркому возможна, но её наблюдают редко.• Лимфосаркомы - внекостномозговые опухоли, состоящие из молодых клеток лимфоидного происхождения - лимфобластов или лимфобластов и пролимфоцитов. Это злокачественные новообразования. Лимфосаркомы и лимфоцитомы могут быть Т- и В-клеточного происхождения. По характеру роста в лимфатических узлах или других очагах выделяют нодулярные и диффузные опухоли. Так же выделяют:• Доброкачественные лимфомы: из малых лимфоцитов; фолликулярная из малых расщеплённых лимфоцитов; фолликулярная смешанно-клеточная; диффузная из малых лимфоцитов (лимфоцитома)• Злокачественные лимфомы: фолликулярная крупноклеточная; диффузная крупноклеточная; диффузная смешанно-клеточная; диффузная иммунобластная (лимфосаркома) • Лимфобластная лимфома (лимфосаркома).• Лимфома Беркитта (л имфосаркома). Группировка на I-IV: I Вовлечение лимфатических узлов одного региона или единичный экстранодальный очаг. II Вовлечение лимфатических узлов двух регионов и более по одну сторону диафрагмы или локализованный экстранодальный очаг по одну сторону диафрагмы .III • Вовлечение лимфатических узлов многих регионов по обе стороны диафрагмы с увеличением селезёнки и/или в сочетании с локализованным экстранодальным поражением .•IV Генерализованное вовлечение в процесс одного или более экстранодальных органов и тканей с увеличением лимфатических узлов или без такого. При поражении печени и костного мозга всегда констатируют IV стадию. Клиническая картина. Клиническая картина лимфоцитом ( доброкачественных лимфом)- опухоль в области лимфатического узла или любой другой локализации, состоящая из зрелых, хорошо дифференцированных лимфоцитов, характер роста чаще всего узловой. Начало болезни зачастую не сопровождается интоксикацией. В крови при лимфоцитомах любой локализации изменения либо отсутствуют, либо выявляют абсолютный лимфоцитоз при невысоком лейкоцитозе, показатели красной крови и тромбоцитов нормальные. Клиническая картина лимфосарком весьма разнообразна - увеличение лимфатических узлов, селезёнки, яичек или доли щитовидной железы; либо предшествовать симптомы интоксикации, развитие аутоиммунной гемолитической анемии, лейкокластического васкулита, полисерозита, экземоподобных высыпаний на коже. Болезнь может начаться с синдрома сдавления венозных и лимфатических сосудов с нарушением функции органов.Для лимфобластной лимфомы типичны быстрое метастазирование в костный мозг и ЦНС, поэтому терапевтический подход аналогичен таковому при остром лимфобластном лейкозе. Диагностика. биопсия опухолевого образования. Показанием к её проведению считают наличие безболезненной плотной опухоли (лимфатического узла, доли щитовидной железы и др.), возникшей без связи с инфекцией. ЛЕЧЕНИЕ 1.Лимфоцитомы. • Во многих случаях полного излечения добиться не удаётся, т.е. проводят паллиативную сдерживающую терапию (циклофосфамид в дозе 100 мг/сут внутрь до стойкого эффекта или хлорамбуцил по 4-6 мг, затем в меньшей поддерживающей дозе). Лечение проводят до получения стабильной полной или частичной ремиссии. Затем возможен перерыв в лечении. При последующих рецидивах аналогичные режимы эффективны в достижении второй, третьей и последующих ремиссий. Резистентность к обычной химиотерапии может свидетельствовать о переходе доброкачественной моноклоновой опухоли в злокачественную. • Хирургическое лечение проводят при локализованных лимфоцитомах, что часто приводит к стойкому многолетнему эффекту • Лучевую терапию проводят в комбинации с полихимиотерапией (используют при локальных лимфоцитомах или при наличии большой опухолевой массы). 2.Лимфосаркомы. Основу лечения лимфосарком составляет многокомпонентная многодневная агрессивная полихимиотерапия. К программам I поколения относят CHOP, BACOP, COMLA, СРОВ. Лечение обычно продолжается 6-8 мес (проводят как минимум 2 курса после констатации полной ремиссии). Частота ремиссий при использовании программ II (COP-BLAM, Pro МАСЕ-МОРР-РгоМАСЕ, mBACOD) и III (МАСОР-В, Pro-MACE-CytaBOM) поколения выше, однако данные по длительной выживаемости при их использовании пока не получены. При резистентных к полихимиотерапии лимфосаркомах рассматривают возможность проведения аутологичной трансплантации костного мозга после подготовки высокими дозами цитостатиков с тотальным облучением тела или без такового. Дальнейшее наблюдение. Контрольное обследование проводят 1 раз в 3 мес на протяжении первых 2 лет, затем каждые полгода, а после 3 лет - 1 раз в год. Обращают внимание на возможность возникновения вторичного лейкоза. После высокодозной химиотерапии нужен более частый контроль. Прогноз. При злокачественных лимфомах современная полихимиотерапия позволяет достичь ремиссии у 70-80% больных; причём 5-летняя выживаемость у них достигает 50-60%. При лимфобластной лимфоме 5-летняя безрецидивная выживаемость достигает 50%, аналогичные показатели получены и при лимфоме Беркитта.
|
|||||||||
|
|
Последнее изменение этой страницы: 2016-08-16; просмотров: 776; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.145.93.221 (0.13 с.) |