Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Ферменты в медицине (ЧАСТЬ ii)
Энзимопатии
Энзимопатии (энзимопатологии) – общее название патологических состояний, развивающихся в результате отсутствия или снижения активности некоторых ферментов. По основной классификации все энзимопатии делят на два типа: 1. Врождённые (наследственные) энзимопатии – заболевания, в основе которых лежит генетически обусловленное отсутствие, остаток или дефектная структура какого-либо фермента. 2. Приобретённые энзимопатии – заболевания, при которых энзимные нарушения наступают вторично, в ходе патологического процесса. Кроме этого, в зависимости от клинического проявления, энзимопатии делят на три группы: 1. Энзимопатии с клиническими проявлениями; 2. Относительно бессимптомные энзимопатии; 3. Бессимптомные энзимопатии. Врождённые (наследственные) энзимопатии
В настоящее время насчитывают около 1500 патологических состояний, начиная от нарушений до тяжёлых состояний с летальным исходом. Частота встречаемости составляет от 1:1000 до 1:1 млн новорожденных. С практической точки зрения наибольшее значение имеют заболевания с летальным исходом, которые встречаются чаще, чем бессимптомные энзимопатии. До настоящего времени нет единой классификации наследственных энзимопатий, но все их делят в зависимости от типа нарушения обмена веществ: 1. Наследственные болезни обмена аминокислот; 2. Наследственные болезни обмена углеводов; 3. Наследственные болезни обмена липидов; 4. Наследственные болезни пуринового и пиримидинового обменов; 5. Наследственные болезни стероидного обмена; 6. Наследственные болезни обмена билирубина; 7. Наследственные болезни обмена металлов; 8. Наследственные болезни порфиринового обмена; 9. Наследственные болезни обмена соединительной ткани; 10. Наследственные болезни обмена крови и кроветворных тканей; 11. Наследственные болезни обмена, обусловленные нарушением (дефектом) канальцев почек; 12. Наследственные болезни обмена, обусловленные дефектом ферментов желудочно-кишечного тракта; 13. Наследственные болезни обмена, вызванные дефектом синтеза белков плазмы и иммуноглобулинов.
Механизм возникновения наследственных энзимопатий
С генетической точки зрения при всех врожденных энзимопатиях синтез энзимов с анормальной структурой и свойствами рассматривается как последствие мутаций. Доказаны мутации только структурных генов, но не исключено существование и мутаций гена-регулятора или гена-оператора. Мутации проявляются характерными изменениями в активности соответствующих энзимов. В одних случаях (у гомозигот по мутантному гену) энзимная активность отсутствует полностью или почти полностью. В других случаях (чаще у гетерозигот) активность энзима снижена незначительно, а в редких случаях даже может быть увеличена. Оценка этих изменений производится путем определения активности энзима в гомогенате некоторых органов (печени) или клетках (эритроциты, лейкоциты) и иногда из сыворотке.
Следует иметь в виду и комплекс факторов, могущих оказать влияние на определяемую активность. Ген-мутант может причинить описанные изменения энзимной активности по нескольким механизмам. 1. Путем синтеза энзима с измененной структурой и измененными каталитическими свойствами. Замена только одной аминокислоты в активном центре энзима может привести к снижению или отсутствию активности вопреки тому, что количество энзимного белка не изменено. 2. Путем синтеза энзима с измененной структурой, неизмененной каталитической активностью и сниженной стабильностью. Энзим денатурируется быстрее и сокращается время его полураспада, из-за чего снижается его активность. 3. Путем изменения скорости синтеза энзимного белка по причине мутации структурного гена или гена-регулятора. При сниженной скорости биосинтеза энзимная активность снижается, при увеличенной – повышается. 4. Путем влияния гена-мутанта на внутриклеточную концентрацию некоторых активаторов или ингибиторов. Эти механизмы нередко комбинируются и это усиливает изменения в энзимной активности. Раскрытие точного механизма измененной энзимной активности при врожденных энзимопатиях часто связано с большими техническими трудностями из-за невозможности изолировать в достаточном количестве и охарактеризовать энзим при низкой активности его у данного индивидуума. Блок обмена веществ
Отсутствие или сильное снижение, активности энзима вызывает прерывание или замедление нормального хода данного пути обмена веществ, что называется блоком обмена веществ (метаболическим блоком). Например, на этапах А, Б, В и Г обмена веществ при врожденной энзимопатии, при которой отсутствует генетический код для синтеза энзима, превращающего В в Г, развивается блок обмена веществ:
В результате такого блока могут проявиться следующие изменения: 1. Концентрация вещества Г сильно снижается, и если это биологически важно, развиваются явления недостаточности. 2. Концентрация вещества В, а иногда и его предшественников Б и А, повышается. При токсичности этих веществ также развиваются симптомы заболевания. Иногда они могут вызвать вторичный блок обмена веществ из-за ингибирования других энзимов. 3. Если вещества А, Б и В не имеют токсического действия, а Г не имеет большого значения для организма, то блок обмена веществ может протекать бессимптомно как доброкачественная аномалия обмена веществ. 4. Концентрация В не увеличивается, если В метаболизируется по другому пути, кроме блокированного. В этих случаях образуются большие количества тех веществ (Д и Е), которые действуют токсически на клетки или ингибируют другие энзимные системы:
5. В случае, если Г может образовываться по другому метаболическому пути, симптомы его недостаточности не проявляются:
Следовательно, для постановки правильного диагноза при врожденных энзимопатиях может быть использовано определение не только активности дефектного энзима, но и концентрации накапливающихся или уменьшающихся метаболитов при блоке обмена веществ.
Примеры наследственных энзимопатий
Типичным примером наследственной энзимопатии, протекающей с клиническими проявлениями является такое заболевание, как фенилкетонурия (ФКУ) или фенилпировиноградная олигофрения. ФКУ – это наследственное заболевание, приводящее в раннем детстве к гибели ребёнка или к развитию тяжёлой умственной отсталости. Частота встречаемости составляет ≈ 1:10000 новорожденных. Молекулярный дефект болезни заключается в блокировании превращения незаменимой аминокислоты фенилаланина (Phe) в тирозин (Tyr) в соответствии с уравнением на рис.5.3.1. То есть, в этом случае организм теряет способность синтезировать фенилаланингидроксилазу. Phe тносится к незаменимым аминокислотам, поскольку ткани животных не обладают способностью синтеза его бензольного кольца. Tyr является полностью заменимым при достаточном поступлении Phe с пищей. В норме избыток Phe поступившего с пищей и неиспользованный для синтеза белка, с помощью фенилаланингидроксилазы превращается в Tyr. У больных ФКУ происходит накопление Phe в тканях и крови примерно в 40 раз выше, чем в норме.
Рис. 5.3.1. Блокирование превращения незаменимой аминокислоты Phe в Tyr
Повышение содержание Phe само по себе не опасно, но стимулирует образование необычных продуктов – производных Phe – фенилпирувата и фенилацетата. Эти вещества оказывают губительное действие на развитие мозга и если не принимать необходимых мер, влекут за собой развитие умственной отсталости. Диагноз ФКУ ставят на основании химического метода открытия Phe и фенилпировиноградной кислоты на пелёнках ребёнка. Для этого используют реактивные бумажки, пропитанные FeCl3 или динитрофенилгидразином. Развитие болезни можно предотвратить, если значительно снизить исключить приём Phe с пищей с самого рождения ребёнка и в течение первых месяцев жизни. За это время мозг успевает окрепнуть, и в дальнейшем патологические продукты обмена Phe уже перестают быть опасны ми. Для такого ребёнка Tyr оказывается незаменимой аминокислотой.
К относительно бессимптомным энзимопатиям относят те случаи, когда отсутствие или недостаточная активность того или иного фермента вызывает болезненные ощущения, но не угрожает жизни человека. Так, например, некоторые люди страдают непереносимостью молока, выражающейся в том, что даже небольшое количество выпитого молока приводит к расстройству кишечника. Причина такой непереносимости заключается в резком недостатке в кишечнике таких людей фермента галактозидазы, расщепляющей молочный сахар. Молочный сахар – это дисахарид лактоза, состоящий из глюкозы и галактозы. Дисахариды в кишечнике не всасываются, они должны быть предварительно разложены на свои составные части – моносахариды. Отсутствие галактозидазы делает невозможным такое разложение лактозы и тем самым препятствует её усвоению. Люди, страдающие непереносимостью молока, легко усваивают молочнокислые продукты (кефир, простоквашу и др.), в ходе приготовления которых молочный сахар разрушается. К бессимптомным энзимопатиям относят те случаи, когда отсутствие фермента изменяет внешность человека (или другие фенотипические признаки), но не нарушает сколько-нибудь важных жизненных процессов. Например, альбинизм характеризуется врождённым отсутствием пигментов в коже, волосах и сетчатке глаза. Метаболический эффект связан с потерей меланоцитами способности синтезировать тирозиназу – фермент, катализирующий окисление тирозина в диоксифенилаланин и диоксифенилаланинхинон, которые являются предшественниками меланина. При этом альбиносы по здоровью не уступают людям с нормальной окраской.
Приобретённые энзимопатии
В качестве примеров приобретённых энзмопатологий можно рассмотреть пищевые и токсические энзимопатии. К пищевым энзимопатиям можно отнести различные авитаминозы, поскольку для работы многих ферментов необходимы витамины, которые являются составной частью фермента. При недостаточном поступлении с пищей того или иного витамина нарушается работа соответствующего фермента. Это, в свою очередь проявляется клинически. Например, такое заболевание как пеллагра связано с отсутствием в пище витамина РР – никотиновой кислоты. Отсутствие или недостаток никотиновой кислоты в пище приводит к нарушению синтеза коферментов дегидрогеназ (NAD и NADF) и соответственно – к нарушению окисления основных субстратов биологического окисления. Наиболее характерными признаками авитаминоза РР, то есть пеллагры являются поражения кожи (дерматиты), поражения желудочнокишечного тракта (диарея) и нарушения нервной деятельности (деменция).
К токсическим энзимопатологиям можно отнести, например, отравление таким ядом, как цианистый калий. Синильная кислота и её соли, в том числе и цианистый калий обладают способностью связывать железо важнейшего дыхательного фермента – цитохромоксидазы. Выход из строя этого фермента приводит к почти мгновенной смерти.
Энзимотерапия
|
||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-04-23; просмотров: 641; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.222.68.81 (0.014 с.) |

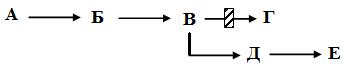

 Фенилаланин
Фенилаланин