Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Модель химической связи в «цифровом» физическом мире.
Как мы излагали выше (7.5), отличительным признаком валентных электронов – а, точнее, атомарных валентных связок «протон-электрон» - является способность к продуцированию зарядовых разбалансов, через сдвиг скважности связующих прерываний в такой связке. Валентный электрон пребывает в достаточно компактной области удержания (6.4), в которой, собственно, на него и действует связующий алгоритм. Хорошо известен также феномен «направленных валентностей» (см., например, [П2]), благодаря которому, связи в сложных молекулах оказываются ориентированы под определёнными углами друг к другу. Это означает, что конфигурация областей удержания валентных электронов в атоме задана достаточно жёстко. Пусть у двух атомов, имеющих по одному валентному электрону, оказались перекрыты их области удержания. Как отмечалось ранее (8.2), электрон, находящийся в зоне такого перекрытия, не может одновременно испытывать действие двух разных алгоритмов, формирующих атомарные валентные связки «протон-электрон». Проще говоря, такой электрон не может входить в состав обоих атомов одновременно. Но при этом мы усматриваем возможности для циклических переформирований составов валентных связок и, соответственно, циклических переключений валентных электронов из состава одного атома в состав другого. В самом деле, пусть валентная связка «протон-электрон» одного из этих атомов приобрела нерезонансную энергию возбуждения, т.е. энергию переменного зарядового разбаланса (7.2). В условиях теплового равновесия, наиболее вероятная частота осцилляций зарядового разбаланса соответствует максимуму равновесного спектра, т.е. энергии 5 kT, где k – постоянная Больцмана, T – абсолютная температура. При наличии у атома энергии квантового возбуждения, согласно нашей модели, работает Навигатор (4.6) – автоматическая программа, которая осуществляет поиск атома-адресата, которому может быть переброшена эта энергия. В качестве атома-адресата выбирается тот, на который расчётная вероятность переброса оказывается максимальной. При тесном соседстве двух атомов, адресатом окажется, с максимальной вероятностью, именно сосед. В свою очередь, этот сосед, приобретший квант энергии возбуждения, перебросит его обратно, и т.д. – два атома, находящиеся в тесном соседстве, будут циклически перебрасывать этот квант энергии друг другу. Если у них не перекрывались бы области удержания валентных электронов, то, при таких перебросах кванта энергии возбуждения, не происходило бы ничего примечательного. Но для случаев перекрытия этих областей, как мы полагаем, приоритеты в управляющих алгоритмах заданы так, чтобы электрон из валентной связки, имеющей колебания зарядового разбаланса, продолжил своё участие в этих колебаниях после их переброса в соседний атом. При этом, конечно, электрон должен переключиться в состав соседнего атома, т.е. две валентные связки «протон-электрон» должны переформироваться.
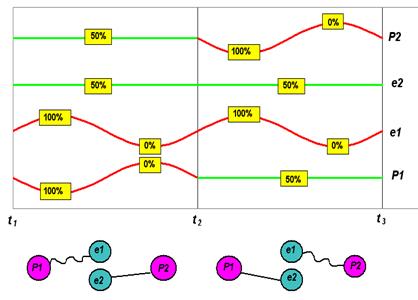
Рис.8.3
Рис.8.3 иллюстрирует результирующие циклические события. Сменяющие друг друга конфигурации валентных связок «протон-электрон» схематически изображены в нижней части рисунка. Волнистая линия, соединяющая протон и электрон, означает наличие колебаний зарядового разбаланса и, соответственно, наличие энергии возбуждения. Период колебаний зарядового разбаланса длится от момента времени t1 до t2, и от t2 до t3; переключения происходят в момент t2. Графики в верхней части рисунка отражают динамику зарядовых разбалансов в валентных связках «протон-электрон». Постоянная 50-процентная скважность связующих прерываний означает отсутствие колебаний зарядового разбаланса, а переменная скважность, с размахом 0-100%, означает наличие этих колебаний. Как можно видеть, оба электрона циклически переключаются из одной валентной связки в другую. Попеременное вхождение электрона в состав того и другого атома как раз и означает наличие динамической сцепки этих атомов. В этом, на наш взгляд, и заключается сущность химической связи. Энергии связи атомарных электронов, через которых атомы сцепляются в молекулы, могут иметь одинаковые или неодинаковые значения – наша модель работает для обоих этих случаев. Разница лишь в том, что, в первом случае, у атомов одинаковы количества периодов связующих прерываний, которые укладываются на одном периоде колебаний зарядового разбаланса, а во втором случае – неодинаковы.
Подчеркнём, что, в отличие от традиционных моделей, дающих статический характер ионной и ковалентной связей, в предлагаемой модели химическая связь имеет динамический характер – будучи циклическим процессом. Чем обеспечивается устойчивость химической связи? На наш взгляд, здесь имеет место развитие принципов, обеспечивающих устойчивость атомных структур. При заданной частоте связующих прерываний в каждой связке «протон-электрон», эти две частицы должны находиться на соответствующем, фиксированном, расстоянии друг от друга (6.4). Если две валентные связки «протон-электрон» химически связаны, то, при небольших увеличениях или уменьшениях расстояния между их протонами, оказывается, что, из-за циклического обмена электронами, длины той и другой валентных связок то несколько больше, то несколько меньше тех, которые должны быть при заданных частотах связующих прерываний. Оба связанных атома, поддерживая «правильные» длины своих валентных связок, в итоге поддерживают «правильную» длину химической связи. А какова энергия химической связи? Наш ответ на этот вопрос кардинально расходится с ортодоксальными воззрениями, где изменения энергии химических связей учитывают в балансах энергий при различных метаморфозах вещества. Заметим, что, при одиночном квантовом перебросе порции энергии с атома на атом, производятся лишь соответствующие перераспределения энергий (4.12) – энергии возбуждения и энергии связи – у этих двух атомов; никакой дополнительной энергией квантовый переброс не обладает. Это должно оставаться верным и тогда, когда квантовые перебросы энергии в паре атомов следуют друг за другом, повторяясь циклически. Соответствующие циклические превращения энергии происходят лишь в этой паре атомов – никак не отражаясь на окружающем мире. Это должно быть справедливо и для самого начала работы вышеописанного циклического процесса, т.е. для момента формирования химической связи: никакого энергетического отклика в окружающем мире при этом не происходит. Тогда мы должны сделать вывод: энергии химических связей, как отдельной формы энергии, не существует. Казалось бы, этот вывод противоречит огромному пласту чуть ли не повседневного опыта. Но мы постараемся показать, что это противоречие – только кажущееся. Во-первых, тепловые эффекты химических реакций, которые считаются бесспорным следствием изменений энергии химических связей, имеют, на наш взгляд, совсем иное происхождение (10.6). Во-вторых, запас устойчивости у стабильной химической связи, конечно, имеет энергетическую меру – энергию диссоциации, при сообщении которой связь разрывается. Но в традиционных методиках определения энергии диссоциации – например, через фотовозбуждение или электронный удар [К4] – разрыв химической связи происходит, на наш взгляд, не оттого, что сообщённая энергия «вытаскивает» пару атомов из потенциальной ямы, глубина которой характеризовала их энергию связи, а оттого, что оказываются невозможны нормальные циклические перебросы энергии возбуждения с атома на атом – а, значит, и циклические переключения электронов из одного атома в другой. Сообщённая здесь «энергия диссоциации» отнюдь не тратится на разрыв химической связи, как таковой, поэтому мы не усматриваем противоречия в том, что для разрыва химической связи требуется энергию «затратить», а при образовании химической связи эта энергия не выделяется.
Заметим, что вывод об отсутствии энергии химических связей вполне согласуется с представлениями о том, что, в отличие от атомных и ядерных структур, напрямую формируемых структуро-образующими алгоритмами (6.4, 6.8), молекулы (за исключением биомолекул в одушевлённых организмах) образуются «сами по себе» - если это допускается физическими параметрами среды (8.1). Энергия структурных связей в веществе, как отдельная форма энергии, имеет место тогда, когда она, по логике «цифрового» физического мира, программно обеспечена: структуро-образующий алгоритм превращает в энергию связи часть энергии в другой форме – например, мы полагаем, что именно так формируется связь на дефекте масс (6.2). Энергия же химических связей программно не обеспечена – поэтому её существование нарушало бы закон сохранения энергии. В самом деле, поразительные свидетельства о том, что «энергия диссоциации» – при сообщении которой химическая связь разрывается – совсем не равна глубине потенциальной ямы, в которой находились связанные атомы, дают молекулярные спектры излучения-поглощения. Для сравнения: в любом связанном состоянии атомарного электрона, его энергия связи всегда равна минимальной энергии, при сообщении которой электрон отрывается от атома. Разумеется, энергии ионизации атома из возбуждённых состояний меньше, чем из основного. Но уровень, на который следует «вытащить» электрон для его отрыва от атома – один для всех связанных состояний. Не наблюдалось случаев, чтобы, при сообщении атомарному электрону энергии, существенно превышающей энергию ионизации из текущего связанного состояния, этот электрон вновь оказывался бы в связанном состоянии. А для молекул подобный феномен нормален – даже в случае одинарной связи. Действительно, справочные значения энергий диссоциации обычно приводятся для основного электронного состояния молекулы – самого сильно связанного [К4]. Но, как следует из молекулярных спектров (см. Рис.8.1, а также, например, [П4]), несколько выше уровня этой первой диссоциации может находиться дно следующего устойчивого электронного состояния, со своим уровнем диссоциации, а выше этого уровня – следующее электронное состояние, и т.д. Нередки ситуации, когда у молекулы, при энергии диссоциации из основного состояния, скажем, 3 эВ, имеются устойчивые электронные состояния, которые выше основного состояния, скажем, на 15 эВ [Т2,Ф4]. Для подобных случаев, пусть сторонники существования энергии химической связи попробуют ответить на вопрос о том, какова же глубина потенциальной ямы, в которой находятся связанные атомы в основном электронном состоянии молекулы – 3 эВ или 15 эВ. Каким образом эта молекула, которая диссоциирует при энергии возбуждения 3 эВ, способна, отнюдь не диссоциируя, поглотить и переизлучить квант в 15 эВ?
В нашей модели этого парадокса нет: если энергия химической связи иллюзорна, то иллюзорны и изменения этой энергии при молекулярном излучении-поглощении. Тогда молекулярные спектры свидетельствуют вовсе не о том, что, при излучении-поглощении квантов молекулой, происходят соответствующие изменения энергии связи атомов. Что же касается ортодоксов, которые энергию химической связи считают реальностью, то они названный парадокс не устраняют и не разрешают – они про него просто помалкивают. Каждому электронному состоянию молекулы ставят в соответствие потенциальную кривую типа потенциала Ми (8.1.1). Считается, что устойчивость молекулы не может быть обеспечена иначе, как с помощью подобной потенциальной ямы – у которой по оси абсцисс отложено межъядерное расстояние. Соответственно, допускаются колебания связанных атомов – около равновесного значения этого расстояния. Полагают, что с помощью квантованных значений энергии таких колебаний объясняется происхождение серий колебательных линий. Но, на наш взгляд, такой подход совершенно неадекватен реалиям. Гладкая и непрерывная кривая потенциальной ямы годится для решения задачи о механических колебаниях – энергия которых зависит от двух параметров, амплитуды и частоты, причём эта энергия отнюдь не квантуется, изменяясь непрерывно. Совсем другое дело – дискретные уровни энергии, переходам между которыми соответствуют кванты, энергии которых зависят не от двух параметров, а только от одного: от частоты. Налицо фундаментальное противоречие: ряды дискретных колебательных энергий молекулы не могут быть обусловлены механическими колебаниями! Но, закрыв глаза на некорректность смешения здесь концепций классических колебаний и квантовых скачков, специалисты проделали огромную работу по согласованию картин колебательных термов и параметров молекулярных потенциальных кривых [Е2]. Так, например, для основного состояния молекулы H2, частота малых собственных колебаний, ~2×1013 Гц, рассчитанная через вторую производную потенциальной кривой ([К4,К2]) и приведённую массу двух атомов водорода, совпадает с частотой, которая соответствует, через постоянную Планка, энергии первого колебательного уровня [Т2,Ф4]. На наш взгляд, подобные результаты подгонок – физически бессмысленны. Ведь теоретики не дали внятных разъяснений – например, для случая той же молекулы H2, имеющей нулевой дипольный момент [Т2] – каким образом при механических вибрациях или ротациях связанной пары электрически нейтральных частиц вещества может поглощаться и излучаться электромагнитная энергия.
Вот почему мы сознательно отказываемся от традиционной модели, согласно которой механические вибрации и ротации молекул имеют отношение к их колебательным и вращательным спектрам. Тайна серий молекулярных линий приоткрывается, если допустить, что в возбуждённой молекуле не происходит ничего, кроме вышеупомянутого циклического процесса перебросов энергии возбуждения с атома на атом – а серии линий молекулярного излучения-поглощения свидетельствуют всего лишь о тех или иных резонансах у этого циклического процесса [Г8].
|
|||||||||
|
|
Последнее изменение этой страницы: 2021-04-04; просмотров: 72; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.14.70.203 (0.019 с.) |