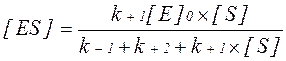Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Коферменти. Простетичні групи
Коферментами називають низькомолекулярні органічні сполуки небілкової природи, що володіють здатністю оборотно зв’язуватися з ферментним білком. Їх можна відділити від апофермента діалізом, дією кислот або лугів, пропусканням через колонку з сефадексом. Утворення молекули ферменту з апо- і коферментів відбувається за рахунок слабих електростатичних або вандерваальсових сил. При цьому змінюється вторинна і третинна структури молекули, після чого фермент-білок стає більш стійким до денатуруючих чинників, ніж апофермент. Коферменти в активному центрі виконують роль переносника різних функціональних груп, протонів і електронів. Окремі коферменти беруть участь в активації молекул субстратів. Коферменти, відщеплюючись від апофермента, здійснюють транспортування цих речовин між окремими ферментами ферментативного ланцюга, перерозподіл їх між органоїдами і гіалоплазмою. В ході каталітичного акту коферменти зазвичай не зазнають глибоких хімічних змін і можуть багато разів (у складі одного або декількох ферментів) брати участь у ферментативних реакціях. Більшість коферментів є похідними вітамінів. Хімічна природа кофермента значною мірою визначає тип і механізми реакцій які вони каталізують. Недостатня кількість вітаміну в раціоні викликає гальмування тих реакцій обміну речовин, в яких бере участь фермент. Найбільш поширені такі коферменти: Нікотінамідаденіндинуклеотид (НАД). Цей кофермент відноситься до універсальних по розповсюдженню і біологічній ролі. Містить каталітично активне угрупування – амід нікотинової кислоти. Структура НАД була вивчена О. Варбургом і X. Ейлером у 1936 р. НАД є складовою частиною понад 40 різних дегідрогеназ:
Сировиною для отримання НАД зазвичай служать дріжджі. Нікотинамідаденіндинуклеотид фосфат (НАДФ). Цей кофермент відрізняється від НАД наявністю третього залишку фосфорної кислоти в положенні 2¢ рибози аденілової частини молекули. НАДФ входить до складу меншого числа ферментів, ніж НАД. НАД в тканинах в 5 – 10 разів більше, ніж НАДФ. НАД і НАДФ є першою ланкою в ланцюзі проміжних переносників водню при біологічному окисленні. Ліпоєва кислота. Є коферментом багатьох оксидоредуктаз. Зв’язується з апоферментом через карбоксильну групу і аміногрупу залишку лізину:
Коферментна функція ліпоєвої кислоти в активному центрі ферменту обумовлена здатністю легко окислюватися і відновлюватися з утворенням дисульфідних зв’язків або сульфгідрильних груп:
Це і дає можливість ферментам, що містять ліпоєву кислоту, брати участь як в перенесенні ацильних груп, так і реакціях біологічного окислення. Глутатіон. Один з найпоширеніших природних пептидів. Трипептид глутатіон утворений із залишків трьох амінокислот: глутамінової, цистеїна і гліцина (g-глутамініл-цистеїніл-гліцин):
Ним багаті печінка, наднирники та еритроцити (75 – 120 мг%). Значення глутатіона в активному центрі ферментів обумовлено наявністю SH-груп, які легко вступають в реакцію окислення з аналогічними групами субстрата або сусідньої ділянки ферменту, утворюючи дисульфідні зв’язки:
В одних випадках це дає можливість ферменту фіксувати субстрат, в інших – одержувати для окисно-відновних реакцій протони і електрони і т.д. Відновлений глутатіон є коферментом деяких ізомераз, що здійснюють перетворення кетоформ кислот в енольні, циc-ізомерів кислот в транс-ізомери та ін. Деякі ферменти (фосфогліцерин-альдегід-дегідрогеназа) містять глутатіон у вигляді простетичної групи. Глутатіон служить коферментом при дії катепсинів, папаїна та ін. Зараз відомо понад 100 ферментів, що містять тіолові групи. Вважають, що глутатіон входить до складу молекули ферменту у відновленій формі. Кофермент А (KoA). KoA – похідне аденілової, фосфорної, пантотенової кислот і b-меркаптоетаноламіна:
Наявність SH-групи у складі молекули кофермента дає йому можливість легко взаємодіяти з жирними кислотами, утворюючи ацилпохідні. KoA іноді називають коферменгом ацилування, або ацетилювання. Відомо понад 70-ти ферментів, у молекулі яких є KoA. Ацетил-КоА бере участь в реакціях перенесення, оксидоредукції, ізомеризації, конденсації і розщепленні ацильних груп. Убіхінони. Ці коферменти, близькі по будові до вітаміну К, є похідними 5-метил-2,3-диметоксіхінона, в молекулі якого до шостого атома вуглецю приєднується поліізопреновий ланцюг (n = 6– 10):
Біологічне значення убіхінонів основане на їх здатності до оборотних окисно-відновних перетворень як проміжних сполук між флавопротеїдами і цитохромами. В ліпідному шарі біологічних мембран їх в 6 – 10 разів більше, ніж цитохромів. 2-Оксоглутарат. Цей метаболіт може виконувати коферментні функції. Складає єдину систему з глутаміновою кислотою в перенесенні аміногрупи трансаміназами:
Нуклеозидфосфати. Ці коферменти беруть участь в реакціях трансфосфорилування і передачі енергії від одного процесу до іншого. Реакції каталізуються кіназами. У ролі коферментів виступають найчастіше такі нуклеозидфосфати: АМФ, АДФ, АТФ, ГМФ, ГДФ, ГТФ, ЦТФ, УМФ, УДФ і інші і, перш за все, АДФ і АТФ. Вони є, з одного боку, джерелами хімічної енергії, яка необхідна для протікання хімічних реакцій, з іншого – складовими частинами багатьох ферментів і коферментів. Зокрема, АТФ є коферментом ряду кіназ, які переносять залишок фосфорної кислоти на монози:
АТФ також може бути джерелом пірофосфата для деяких вуглеводів:
АТФ часто виступає в ролі кофермента при перенесенні аденозинмонофосфорного залишку на яку-небудь кислоту, зокрема, на амінокислоту:
АТФ – джерело аденозильного залишку при активації іперенесенні метильних груп. Як коферменти часто виступають уридинфосфати і їх похідні. Наприклад, УДФГ використовується при активації, перенесенні і перетворенні цукрів: ізомеризації галактози в глюкозу, синтезі сахарози, лактози, глюкозидів, хітину і т.д. Коферментні функції виконують і інші нуклеозидфосфати. Зокрема, з участю цитидинфосфатів протікають реакції синтезу окремих видів фосфатидів, особливо лецитину. Тетрагідрофолієва кислота (ТГФК). Це гомолог фолієвої кислоти, у якої відновлені атоми водню і азоту в положеннях 5, 6, 7 і 8:
ТГФК є коферментом великої групи трансфераз, що каталізують реакції, пов’язані з перенесенням або активацією одновуглецевих залишків: –СОН, –СН2–ОН, –СН3. З участю ТГФК синтезуються і розщеплюються пуринові основи і деякі амінокислоти: гістидин, гліцин і, особливо, серин. B12-залежні коферменти. Ці коферменти є похідними вітаміну В12 в молекулі якого ціаниста група заміщена на 5¢-дезоксиаденозин. Кофермент B12 разом з тетрагідрофолієвою кислотою бере участь в перенесенні метильних груп. Під впливом відповідної метилтрансферази від 5-метилтетрагідрофолієвої кислоти метильна група переноситься на кофермент B12 і приєднується до його молекули замість 5¢-дезоксиаденозина. Потім під впливом іншої трансферази ця ж метильна група переноситься на речовину, яка необхідна для синтезу метилвмісної сполуки. Ним може бути гомоцистеїн, з якого утворюється амінокислота метіонін. Після метилування вітамін B12 знову перетворюється в кофермент B12 і вступає в реакцію з новими порціями 5-метилтетрагідрофолієвої кислоти. Крім того, кофермент B12 бере участь в найважливішій для біосинтезу жирних кислот реакції – ізомеризації сукциніл-КоА в метилмалоніл-КоА:
Кофермент B12 бере участь в ізомеризації глутамінової кислоти, перетворенні діолів у альдегіди та в інших важливих реакціях обміну речовин. Простетичні групи. Багато ферментів у складі своїх молекул містять кофактори, які міцно зв’язані ковалентними зв’язками з апоферментами. Молекули таких ферментів слабо або зовсім не дисоціюють. При утворенні молекули ферменту з апофермента і простетичної групи утворюється стійка вторинна і третинна структура. З кофакторів такого роду найбільший інтерес представляють такі:
Флавіннуклеотиди. Флавіннуклеотиди – це похідні вітаміну B2, або рибофлавіну. В тваринних клітинах зустрічаються в основному флавінмононуклеотиди (ФМН) і флавінаденіндинуклеотиди (ФАД):
ФМН і ФАД – простетичні групи флавінових ферментів, або флавопротеїдів. Зокрема, ФМН як простетична група, з’єднуючись з апоферментом, утворює холофермент – „жовтий дихальний фермент” (ЖДФ):
Ізоалоксазонове угрупування простетичної групи дає можливість ЖДФ виконувати найважливіші каталітичні функції, зокрема, брати участь в реакціях біологічного окислення. ЖДФ є переносником протонів і електронів в двох напрямах: у бік цитохромів або від цитохромів до піридиннуклеотидів дихального ланцюга і до окислених субстратів. Крім того, флавіннуклеотиди беруть участь у ряді інших реакцій обміну речовин. Серед них особливе місце займають реакції утворення подвійних зв’язків. Зокрема, сукцинатдегідрогеназа і ацил-КоА-дегідрогеназа циклу b-окислення жирних кислот каталізують процеси, пов’язані з утворенням подвійних зв’язків:
Тіамінфосфати. Тіамінфосфати є похідними вітаміну В1 або тіаміну. Простетичною групою ферментів найчастіше виступає тіамінпірофосфат (ТПФ). З’єднання тіамінфосфатів з апоферментами, мабуть, таке ж, як і флавіннуклеотидів. Каталітична активність ферменту пов’язана з іонізацією вуглецевого атома в положенні 2 тіазолового кільця, до яких і приєднується молекула субстрата:
Під впливом позитивного заряду атома азоту від вуглецевого атома в положенні 2 відщеплюється протон, виникає негативно заряджений карбаніон. Карбаніон – нуклеофільний центр, який легко вступає в реакцію з субстратом, наприклад, з піровиноградною кислотою, утворюючи піруват-ТПФ-комплекс. Цей комплекс легко декарбоксилується, утворюючи альдегідо- або кето-ТПФ-комплекс, карбонільна група якого передається на відповідний акцептор, наприклад, на ліпоєву кислоту. ТПФ після цього вступає в реакцію з новими порціями субстрата. ТПФ є простетичною групою не тільки піруватдекарбоксилази, але й інших ферментів, що беруть участь в обміні багатьох a-кетокислот, утворенні і розщепленні кетоцукрів, a-оксикетонів і дикетонів.
Піридоксальфосфат і піридоксамінфосфат – похідні вітаміну В6. Є складовою частиною піридоксалевих ферментів. Значення цих ферментів у білковому обміні велике, оскільки з їх діяльністю пов’язана більшість перетворень a-амінокислот: переамінування, декарбоксилування, рацемація, конденсація b- і g-заміщених a-амінокислот і т.д. Теорія каталітичної дії цих ферментів вперше була розроблена радянськими біохіміками О.E. Браунштейном і M.M. Шемякіним на прикладі реакцій переамінування:
Ці реакції дають можливість організму отримати потрібні замінні амінокислоти для клітин і тканин. Крім того, окремі декарбоксилази, що містять в своїх молекулах піридоксальфосфат або піридоксамінфосфат, каталізують утворення в тканинах фізіологічно активних амінів: гістаміна з гістидина, серотоніна з триптофана та ін. З діяльністю піридоксалевих ферментів пов’язано утворення цистеїна з серина і гомоцистеїна, тирозина, триптофана. Біотин. Приєднання біотина до апоферменту здійснюється за допомогою пептидного зв’язку, який утворюється за рахунок карбоксильної групи біотина і e-аміногрупи залишку лізину простого білка. Біотин входить до складу молекул багатьох ферментів, що каталізують реакції карбоксилування і перенесення карбоксильних груп. Залізопорфіринові комплекси є простетичною групою каталази, пероксидази і цитохромів. У каталазі і пероксидазі, як і в гемоглобіні крові, простетична група – гем. Залежно від природи гема цитохроми ділять на чотири групи: 1. Цитохроми А – містять залізоформілпорфірин. 2. Цитохроми В – містять залізопротопорфірин. 3. Цитохроми С – містять заміщений залізомезопорфірин з ковалентними зв’язками між білком і порфірином. 4. Цитохроми D містять залізодигідропорфірин. Апоферменти – низькомолекулярні білки. Кожна група цитохромів складається з індивідуальних представників. Вони позначаються малими латинськими буквами і цифровими підрядковими індексами: а, a1, а2, в, в1, в2 і т.д. Зв’язок між апоферментом і простетичною групою найчастіше ковалентний і додатковий. Прикладом може бути цитохром с:
Цитохроми є складовою частиною дихального ланцюга. В ході функціонування якого електрони послідовно переносяться від „жовтого дихального ферменту” на цитохром b, c1, c, а і а3. Під впливом ферменту, цитохромоксидази цитохром а окислюється, електрон, що відщепився, переноситься на атом кисню, який іонізується і реагує з іоном водню. Утворюється вода. Енергія акумулюється у вигляді АТФ. Ізоферменти Ізоферменти – це різновиди ферменту, які володіють однією і тією ж субстратною специфічністю, але відрізняються між собою деякими фізичними, хімічними, каталітичними та імунологічними властивостями. Молекула ізофермента складається з декількох субодиниць, які формують її четвертну структуру. В даний час відомі ізоферменти декількох ферментів: лактатдегідрогенази (ЛДГ), фосфоглюкомутази, альдолази, креатинкінази, малатдегідрогенази, ізоцитратдегідрогенази, глюкозо-6-фосфатдегідрогенази, фосфоглюконатдегідрогенази та ін.
Найбільш детально вивчені ізоферменти ЛДГ, яка здійснює перетворення молочної і піровиноградної кислот. Молекулярна маса ЛДГ складає 140 тис. Її молекула складається з чотирьох субодиниць, які діляться на два види: H (переважають в міокарді) і M (найбільше в скелетній мускулатурі). Для ЛДГ тварин характерні п’ять ізоферментів, що складаються з таких субодиниць: I – HHHH, II – HHHM, III – HHMM, IV – HMMM і V –MMMM. Ізоферменти мають нумерацію: I – ЛДГ1 II – ЛДГ2, III – ЛДГ3, IV – ЛДГ4 і V – ЛДГ5. Перші два ізофермента (ЛДГ1 і ЛДГ2) при електрофорезі рухаються до анода – анодні фракції, останні два (ЛДГ4 і ЛДГ5) рухаються до катода – катодні фракції і середній ізофермент (ЛДГ3) займає проміжне положення. Фракції відрізняються між собою біологічною роллю. Так, анодні фракції здійснюють процес перетворення піровиноградної кислоти в аеробних умовах, катодні – в анаеробних, а проміжна фракція – в аеробних і анаеробних. Для кожної тканини і органу характерний свій ізоферментний спектр. Так, в міокарді, мозку, нирках і еритроцитах переважають ізоферменти ЛДГ1 і ЛДГ2. Саме під їх впливом з молочної кислоти утворюється піровиноградна кислота, яка служить джерелом ацетил-КоА. ЛДГ4 і ЛДГ5 типові для скелетної мускулатури, тканин легень і печінки. Під впливом цих ізоферментів піровиноградна кислота відновлюється до молочної кислоти. ЛДГ3 переважає в тканинах ендокринних залоз і селезінки. Спектр ізоферментів може змінюватися в різні періоди онтогенезу і при патології. Його визначення дає можливість діагностувати хворобу і контролювати лікування. Так, при нефриті у складі крові і сечі з’являються додаткові фракції ЛДГ4 і ЛДГ5, нетипові для здорових тварин. При захворюваннях легень у крові і сечі зростає вміст ЛДГ3. Механізм дії ферментів Механізм ферментативного каталізу є об’єктом дослідження багатьох учених. При ферментативному каталізі виявляється білкова природа ферментів, їх термолабільність, вплив рН середовища, специфічність дії, висока каталітична здатність, чутливість до активаторів і інгібіторів. Ферментативна реакція протікає згідно закону дії мас. Ферменти і енергія активації. Ферменти – це біологічні каталізатори. Вони впливають на швидкість реакцій в двох напрямах: прискорюють розщеплення або синтез певних речовин. Збільшення швидкості реакції відбувається внаслідок зміни енергії активації молекул субстратів. Енергія активації E характеризує енергетичний бар’єр, який необхідно подолати, щоб привести реагуючі речовини в активний стан. Він обумовлений силами міжмолекулярного зчеплення або міжмолекулярного відштовхування реагуючих речовин. Швидкість реакції можна збільшити, якщо збільшити число молекул, що активуються, або зменшити висоту енергетичного бар’єру. Молекули субстратів можна привести в активний стан різними прийомами. Одним з них є нагрівання, при якому в розчинах зростає швидкість руху молекул і можливість їх зіткнення. За наявності каталізатора знижується енергетичний бар’єр і енергія активації. Так, для розщеплення сахарози на глюкозу і фруктозу під впливом сірчаної кислоти E= 134400 Дж/моль, а за участю інвертази – 40420 Дж/моль. Для гідролізу казеїну соляною кислотою E =109200 Дж/моль, пепсином – 50400 Дж/моль. Енергія активації розкладання Н2О2 без каталізатора рівна 75600 Дж/моль, з участю колоїдної платини – 49140, каталази печінки – 23100 Дж/моль. Теорія ферментативного каталізу. Існуючі теорії, пояснюючи взаємодію ферменту і субстрата, допускають тимчасове їх з’єднання з утворенням проміжного фермент-субстратного комплексу. Теорія ферментативного каталізу (теорія Міхаеліса – Ментен) припускає такі етапи: I етап. Між субстратом і ферментом виникає зв’язок, внаслідок чого утворюється фермент-субстратний комплекс ES, в якому компоненти зв’язані між собою ковалентним, іонним, водневим та іншими зв’язками. II етап. Субстрат під впливом приєднаного ферменту активується, стаючи доступним для відповідних реакцій каталізу ES. III етап. Здійснюється каталіз ES*. IV етап. Вивільнюється молекула ферменту E і продукти реакції P. Послідовність перетворень відображає схема:
Теорія ферментативного каталізу була підтверджена експериментально. Так, з хріну був виділений фермент, що розщеплює пероксид водню – пероксидаза коричневого кольору. Після з’єднання ферменту Е з субстратом Н2O2 (S) виникає фермент-субстратний комплекс ES зеленого кольору. Через деякий час субстрат активується, утворюючи фермент-активований субстрат ES* червонуватого кольору. Він розщеплюється на коричневий фермент E і продукти розпаду P. Б. Чанс розробив чутливі методи для спектрофотометричного вивчення кінетики ферментативних реакцій на всіх етапах каталізу. Кінетика ферментативних реакцій. Кожна хімічна реакція протікає з певною швидкістю. Біологічне призначення ферментів полягає в направленому підвищенні швидкості хімічних реакцій. Ферментативна кінетика – розділ хімічної кінетики, що вивчає залежність швидкостей ферментативних реакцій від хімічної природи реагуючих речовин (субстратів, ферментів) і умов їх взаємодії (концентрації компонентів, рН, складу середовища, температури, дії активаторів або інгібіторів та ін.). Розрізняють декілька типів ферментативних реакції: необоротні реакції з одним субстратом, оборотні реакції з одним субстратом, необоротні реакції з двома субстратами і т.д. Найбільш поширені необоротні реакції з одним субстратом. Згідно теорії ферментативного каталізу (Л. Міхаеліс і M. Ментен; Дж. Бріггс і Дж. Холдейн), фермент E спочатку реагує з субстратом S, що призводить до утворення фермент-субстратного комплексу ES (в багатьох реакціях – двох і більше комплексів, що виникають в певній послідовності). Фермент-субстратний комплекс характеризується константою швидкості реакції його утворення k+1 і константою швидкості реакції розпаду k–1:
Для характеристики утворення фермент-субстратного комплексу була прийнята субстратна константа, або константа дисоціації Кs, яка рівна:
Величина субстратної константи залежить від природи субстрату і ферменту. При однакових початкових концентраціях ферменту і субстрату концентрація комплексу [ES] буде тим більша, чим менша величина Кs:
Субстратна константа визначає ступінь спорідненості ферменту і субстрату. Так, для інвертази, яка здійснює гідролітичне розщеплення сахарози до глюкози і фруктози, KS= 0,0167. Тут концентрація фермент-субстратного комплексу перевищує концентрацію вільних ферменту і субстрата приблизно в 60 разів. У другій фазі ферментативного каталізу фермент-субстратний комплекс розпадається на фермент E і продукт реакції P. Обидві фази ферментативного каталізу з’єднано в систему, типову для необоротних реакцій з одним субстратом:
В системі k+2 означає константу швидкості розпаду фермент-субстратного комплексу на фермент Е і продукт реакції P. Для повної характеристики ферментативного процесу використовується константа Міхаеліса Кm. Вона виражає відношення констант трьох реакцій, показаних в системі:
Числовий вираз Кm завжди дещо більше, ніж Ks. Так, значення Кs для фермент-субстратного комплексу сахараза – сахароза рівно 0,0167, а величина Кm – 0,0280 моль/л. Кm для різних ферментів неоднакова. Швидкість хімічної реакції, що каталізується ферментом, вимірюється кількістю молів субстрата, які перетворюються за одиницю часу. Так, швидкість ферментативної реакції, що протікає за оптимальних умов (повне насичення ферменту субстратом, температура 25°С або 37°C, рН), позначається символом V і називається максимальною, або початковою, швидкістю. Вона визначається константою швидкості розпаду фермент-субстратного комплексу і концентрацією ферменту:
Для ферментативної реакції, яка протікає при недостатньому насиченні ферменту субстратом, характерна спостережувана швидкість
Якщо врахувати, що
і
то
Таким чином, швидкість ферментативної реакції прямо пропорційна константі швидкості розпаду фермент-субстратного комплексу з утворенням продукту реакції, концентраціям ферменту, субстрату і обернено пропорційна субстратній константі. Для стаціонарної стадії реакції концентрація [ES] постійна. Якщо [S] > [Е],то
Застосовуючи константу Міхаеліса, можна спростити рівняння:
Спостережувану швидкість реакції можна виразити рівнянням:
При збільшенні концентрації субстрату [S], коли [S] > Km
Тут швидкість ферментативної реакції прагне до максимальної швидкості. Швидкість реакції можна охарактеризувати рівнянням Міхаеліса – Ментен:
З рівняння витікає, що при незначній концентрації субстрату швидкість ферментативної реакції лінійно залежить від [S], а при дуже високій концентрації субстрату вона прагне максимальної швидкості Vmax і вже не змінюється із збільшенням [S]. Рівняння також показує, що при збереженні рівня [S] > [E] швидкість реакції пропорційна концентрації ферменту E.
|
|||||||||
|
|
Последнее изменение этой страницы: 2017-02-07; просмотров: 217; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.21.231.245 (0.062 с.) |




















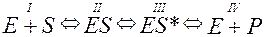
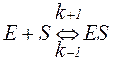





 . Вона визначається добутком константи швидкості розпаду фермент-субстратного комплексу з утворенням продукту реакції Р і концентрації фермент-субстратного комплексу [ES]:
. Вона визначається добутком константи швидкості розпаду фермент-субстратного комплексу з утворенням продукту реакції Р і концентрації фермент-субстратного комплексу [ES]:
 ,
,