
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Особливості фізико-хімічних (інструментальних)Стр 1 из 17Следующая ⇒
Особливості фізико-хімічних (інструментальних) Методів аналізу
Основне завдання аналітичної хімії - встановлення якісного та кількісного складу речовин, а також ідентифікація та встановлення будови молекул нових речовин. Класичні методи аналітичної хімії (хімічні методи аналізу) - гравіметричний та титриметричний аналіз - дозволяють визначати якісний компонентний склад речовин з межею виявлення 10-3 - 10-5 %. Діапазон кількісного визначення концентрацій компонентів складає 0,01 – 100 % при відносній точності результатів аналізу 0,2 %. Хімічні методи аналізу характеризуються використанням простого обладнання але вимагають застосування великої кількості ручних операцій і тривають довгий час (від десятків хвилин до декількох годин), слабко піддаються автоматизації. Кількісний аналіз речовин, вміст яких не перевищує 0,01 %, практично неможливий. Такі показники хімічних методів аналізу не задовольняють запити сучасної хімічної промисловості та промисловості виробництва матеріалів для будівництва, сільського господарства, радіоелектроніки, космічної техніки, атомної енергетики, медицини, наукових досліджень. Перед аналітичною хімією стоїть завдання розробки методів аналізу з межею виявлення 10-5 - 10-10 % і нижче в процесах виробництва надчистих матеріалів. При аналізі об'єктів навколишнього середовища і екологічному контролі діючих виробництв необхідно проведення аналізу великої кількості проб повітря, стічних вод, відходів виробництва. Це вимагає розробки експресних автоматизованих методик аналізу. Інколи виникає необхідність аналізувати об'єкт без руйнування (без відбирання проби) або визначати не середній склад, а склад в деяких точках на поверхні чи в об'ємі об'єкта (локальний аналіз). Цим вимогам найбільш повно відповідають фізико-хімічні методи аналізу (ФХМА). Відмінність ФХМА від хімічних методів полягає в тому, що для одержання видимого аналітичного сигналу використовують прилади, які перетворюють яку-небудь властивість хімічної системи в переважно електричний сигнал, який легко зареєструвати вимірювальними приладами або записати у вигляді графіків на паперових носіях чи дисплеях. Фізико-хімічні методи аналізу поділяються на 2 групи:
1. Власне фізико-хімічні методи, які грунтуються на вимірюванні фізичних або фізико-хімічних властивостей (параметрів) системи при проведенні хімічної реакції з об'єктом аналізу. 2. Фізичні методи аналізу, які грунтуються на вимірюванні фізичних властивостей (параметрів) системи без проведення хімічних реакцій. Між цими групами методів чіткої границі немає, об'єднує їх те, що аналітичний сигнал вимірюється за допомогою приладів (інструментів), тому інколи ці методи називаються інструментальними. Класифікація ФХМА основана на спільності теоретичних і практичних принципів одержання аналітичного сигналу. Загальне число ФХМА перевищує декілька десятків, але найбільш поширені такі: 1. хроматографічні методи аналізу; 2. спектральні (оптичні) методи; 3. електрохімічні методи; 4. радіометричні методи; 5. масспектрометричні методи; 6. рентгеноспектральні методи. Крім методів, в яких використовуються прилади, що грунтуються на одному принципі, існують методи, в яких використовуються різні принципи для одержання аналітичного сигналу. Такі методи називаються гібридними. Майже у всіх ФХМА для встановлення складу речовини використовується 2 методичних засоби: метод прямих вимірювань і метод титрування. У загальному вигляді, процес одержання даних про хімічний склад об'єкта аналізу в прямих методах складається з таких етапів (рис. 1.1):
Рис. 1.1. Загальна схема проведення аналізу
1. Відбір проби. Об'єкти аналізу можуть бути тверді, сипучі, рідкі, газоподібні речовини або суміші, які знаходяться в різних місткостях, в яких вони зберігаються або транспортуються, а також у трубопроводах. Маса таких об'єктів може складати кілограми і навіть тони. Для того, щоб одержати інформацію про склад об'єкта аналізу, необхідно відібрати порівняно невелику пробу (10-100г) і доставити її в аналітичну лабораторію, яка є на будь-якому хімічному виробництві. Проба повинна бути представницькою, тобто склад і властивості її повинні відповідати середньому складу і властивостям об'єкта аналізу. Існують певні правила відбору представницьких проб у залежності від характеру і агрегатного стану об'єкта, які обумовлюються державними стандартами. Цих правил необхідно дотримуватися.
2. Обробка проби. Перед аналізом проба піддається обробці. Це можуть бути фізичні, хімічні, механічні та інші процеси: подрібнення, розчинення, виділення з проби визначуваних компонентів, переведення в іншу хімічну сполуку, відокремлення компонентів, які заважають визначенню, тощо. 3. Одержання аналітичного сигнала. Аналітичний сигнал – це кількісна характеристика, величина якої пов'язана з хімічним складом аналізованої речовини. У хімічному аналізі підготована проба піддається дії певних реактивів і нескладних хімічних операцій (відокремлення осадів, висушування, зважування, визначення об'єму тощо). Аналітичний сигнал одержують у вигляді видимого результату перебігу хімічної реакції: утворення осаду, забарвлення розчину, маси речовини точно відомого складу, об'єму титранта. Аналітичний сигнал фізико-хімічних методів аналізу одержують за допомогою приладів. Це можуть бути: сила струму, потенціал, інтенсивність випромінювання або поглинання світла (однопараметрові сигнали), а також їх залежність від часу, об'єму розчину, довжини хвилі (двопараметрові сигнали). Можуть бути навіть трипараметрові сигнали. Чим більша розмірність сигналу, тим більша його інформативність, але тим складніший прилад. Якщо при певних умовах який-небудь параметр аналітичного сигнала залежить від виду досліджуваної речовини і не залежить від його вмісту в пробі, такий параметр сигнала може бути використаний для якісного аналізу. Для кількісного аналізу використовується параметр аналітичного сигнала, який залежить від кількості або від концентрації речовини. 4. Обробка аналітичного сигнала. Для одержання результату аналізу аналітичний сигнал відповідним чином необхідно обробити. В хімічних методах якісного аналізу візуальне спостереження аналітичного сигнала дозволяє зразу зробити висновок про наявність або відсутність певної речовини. В кількісному хімічному аналізі результати аналізу розраховують за нескладними формулами, використовуючи величини виміряні в п.3. У ФХМА величина параметрів аналітичного сигнала залежить не тільки від хімічного складу проби, але і від низки параметрів, при яких проводяться процеси 2 і 3. Частина цих параметрів (ui) контролюється і природа їх впливу на аналітичний сигнал відома. Інша частина (vi) не контролюється і природа їх дії може бути не відома.
До контрольованих параметрів можуть належати температура, pH середовища, об'єм або маса проби, тощо. Неконтрольованими параметрами можуть бути наявність невідомих домішок, зміна характеристики самого вимірювального приладу, тощо. Неконтрольовані параметри зумовлюють випадкові похибки результату аналізу і визначають його відтворюваність. Обробка аналітичного сигнала у ФХМА полягає в перетворенні показника прилада y в результат аналізу x. Незважаючи на відому функціональну залежність аналітичного сигнала y від визначуваної величини x та параметрів контрольованих умов ui, аналітичним розв'язком рівняння (1.1) відносно x (x = j (y, ui)) практично не користуються через складність точного врахування впливу всіх можливих параметрів. Як правило, реальна функціональна залежність аналітичного сигнала від визначуваної величини у кожному окремому випадку знаходиться експериментально.
Процес експериментального визначення залежності параметру аналітичного сигнала від складу проби називається калібруванням (градуюванням). Калібрування здійснюється за допомогою комплекту стандартів або еталонів - сумішей або зразків з відомим вмістом одного чи декількох визначуваних речовин. Основні вимоги до комплекту стандартів: однаковий агрегатний стан з обробленим об'єктом аналізу, який поступає в прилад; близькість складу невизначуваних компонентів (матриці) стандарта і зразка; очікуваний вміст компоненту в зразках повинен знаходитися в межах вмісту визначуваних компонентів в комплекті стандартів. Стандарти можуть виготовлятися в хімічних лабораторіях з чистих реактивів, які відповідають вимогам, що ставляться до речовин первинних стандартів. Склад таких стандартів розраховують, виходячи з точної кількості взятих речовин-стандартів і точної кількості або об'єму стандартів. Інколи комплекти еталонів виготовляють спеціалізовані організації (особливо, якщо об'єкт аналізу твердий або сипучий матеріал). У цьому випадку до комплекту еталонів додається паспорт, який містить відомості про склад окремих еталонів і рекомендації по його застосуванню. У залежності від конкретних умов одержання аналітичного сигнала використовуються різні методи калібрування (градуювання). 1. Метод прямого або абсолютного калібрування. Цей метод використовується тоді, коли конкретний метод ФХМА дозволяє при одержанні аналітичного сигнала від зразків і еталонів (етап 3) підтримувати постійними в часі величини контрольованих параметрів. Тоді функціональна залежність аналітичного сигнала буде мати вигляд: y = j (x), ui = const, (1. 2) і з допомогою комплекту стандартів можна зняти функціональну залежність параметра аналітичного сигнала (yj) від вмісту визначуваного компонента в стандартах (xj, де j - порядковий номер еталона). Цю залежність будують у вигляді графіка в координатах y - x (калібрувальний графік), або виражають в аналітичному вигляді за допомогою математичних методів (калібрувальна функція). Аналітичний сигнал від досліджуваного зразка одержують при тих же значеннях контрольованих умов і, користуючись калібрувальним графіком або функцією, за виміряним значенням y визначають параметр x (рис 1.2.).
Рис. 1.2. Побудова калібрувального графіка і визначення результата аналізу в методі прямого (абсолютного) калібрування.
Метод простий і достатньо точний в залежності від того, з якою точністю виконується умова ui = const. Для зменшення похибок, пов'язаних з можливими змінами неконтрольованих параметрів, необхідно проводити процедуру калібрування перед кожною серією аналізів досліджуваних зразків. Різновидом метода прямого калібрування є метод калібрувальних коефіцієнтів. Досить часто калібрувальний графік є прямою лінією, що проходить через початок координат. Така залежність має простий вигляд x=kּy. Якщо ця залежність достовірно встановлена за допомогою відповідного комплекту стандартів, немає потреби кожен раз проводити калібрування з використанням всього комплекту. Калібрувальний коефіцієнт можна розрахувати, вимірявши аналітичний сигнал від одного стандарта з більшим вмістом визначуваного компонента:
За точністю цей метод не поступається першому, але простіший у використанні. Тому намагаються проводити 3-й етап аналізу в таких умовах, щоб виконувалася пряма пропорційна залежність аналітичного сигнала від концентрації або маси визначуваної речовини у зразку. 2. Метод відносного калібрування або метод внутрішнього стандарта використовується тоді, коли з тих чи інших причин не забезпечується умова постійності в часі контрольованих параметрів (ui ≠ const). Цей метод полягає у тому, що до об'єкта аналізу і еталонів додають постійну кількість речовини-стандарта (внутрішнього стандарта), якої немає в об'єкті аналізу. Вибирають таку речовину і таку її кількість, щоб вплив контрольованих умов на вимірюваний параметр аналітичного сигнала визначуваної речовини і речовини-стандарта був однаковим. Це є необхідною умовою для використання метода відносного калібрування. Однаковий вплив контрольованих умов повинен реалізуватися так, щоб функціональна залежність параметра аналітичного сигналу дорівнювала [PS1] добутку або сумі двох функцій, одна з яких є функція, яка залежить від вмісту визначуваної речовини, друга - від контрольованих умов: 1. 2. У першому випадку відношення параметрів аналітичного сигнала визначуваної речовини і речовини стандарта не буде залежати від параметрів роботи приладу (ui). При постійному вмісті речовини-стандарта в еталонах і зразках (xст = const) відносний параметр аналітичного сигнала r = y/yст буде залежати тільки від вмісту визначуваної речовини в зразках:
У другому випадку відносний параметр аналітичного сигнала дорівнює різниці аналітичних сигналів визначуваної речовини і речовини-стандарта:
Результат аналізу одержують, як в пункті 1, використовуючи залежність відносного параметру аналітичного сигнала r від визначуваної величини x. Цей метод обробки аналітичного сигнала більш трудомісткий ніж метод абсолютного калібрування, бо вимагає додаткової операції додавання речовини внутрішнього стандарта і одночасного вимірювання двох аналітичних сигналів з наступним розрахунком відносного аналітичного сигнала, але дозволяє при нестабільності контрольованих умов одержати результати аналізу, близькі за точністю до метода абсолютного калібрування.
Якщо об'єкт аналізу становить основну речовину з невеликою кількістю домішок, при визначенні кількості домішок методом відносного калібрування за речовину-стандарт доцільно взяти основний компонент зразка, концентрація якого змінюється незначно і може вважатися постійною. У цьому випадку аналіз спрощується за рахунок відсутності операції спеціального додавання речовини-стандарта. Різновидом методу внутрішнього стандарту є метод добавок. У цьому методі як речовину-стандарт беруть визначувану речовину, відому кількість якої додають до відміряної кількості об'єкта аналізу. Процедура методу полягає в тому, що спочатку вимірюють аналітичний сигнал від об'єкта аналізу. Потім проводять серію вимірювань аналітичного сигналу від сумішей об'єкта аналізу з різними відомими добавками чистої визначуваної речовини або її розчину відомої концентрації. Екстраполяція залежності аналітичного сигналу від кількості добавленої речовини до нульового значення аналітичного сигналу (рис. 1.3.) дає можливість визначити вміст аналізованої речовини в зразку. Оскільки при нелінійності калібрувальної функції екстраполяція може привести до значної помилки результату аналізу, звичайно цей метод використовують при прямій пропорційній залежності аналітичного сигналу. У цьому випадку точність методу задовільна і можна використати аналітичний розрахунок результату аналізу.
Рис. 1.3. Визначення результату аналізу методом добавок.
Припустимо, що аналітичний сигнал від проби з концентрацією сx є yx. При лінійній залежності:
Якщо підготовку проби організувати так, щоб після додавання чистої речовини концентрація визначуваної речовини в пробі збільшилася на сст, одержимо:
З цих двох рівнянь можна визначити результат аналізу:
Якщо до зразка об'єму Vx додають стандартний розчин об'ємом Vст з концентрацією речовини сст і одержують аналітичний сигнал yx+ст, результат аналізу розраховують за формулою:
яка враховує зменшення концентрації визначуваної речовини від збільшення об'єму внаслідок додавання стандарту. Для забезпеченняя однакових умов впливу складу матриці на аналітичний сигнал необхідно, щоб об'єм стандарта був набагато меншим ніж об'єм проби (Vст «Vx). Якщо ця умова виконується, то з достатньою точністю можна користуватися спрощеною формулою:
Звичайно намагаються готувати стандарт з такою концентрацією сст, щоб при обраному об'ємі стандарту (Vст), аналітичний сигнал збільшувався приблизно вдвоє, тобто yx+ст ~ 2yx. Метод добавок дозволяє одержувати достатньо точні результати при концентраціях речовин, які лежать на границі межі виявлення. У методах титрування ФХМА використовуються для фіксування точки еквівалентності. Під час титрування вимірюється інтенсивність аналітичного сигналу в залежності від об'єму титранта. Вигляд кривих титрування може бути різний через те, що інтенсивність аналітичного сигналу по-різному залежить від концентрації визначуваної речовини, титранта чи продукту реакції. Для визначення точки еквівалентності необхідно вибрати для вимірювання такий параметр розчину, щоб на кривій титрування у точці еквівалентності був помітний стрибок, злам або різка зміна кута нахилу прямих чи дотичних до кривих. Для точнішого фіксування точки еквівалентності, інколи будують залежність від об'єму титранту не самого аналітичного сигналу, а його першої або другої похідної, що легко організувати апаратним шляхом. Одержання правильних результатів аналізу можливе тільки при виборі відповідного способу калібрування. Для цього необхідно бути добре обізнаним з теоретичними засадами фізичних або фізико-хімічних явищ, які покладені в основу методу.
Питання для самоконтролю. 1. Недоліки (обмеженість) хімічнихх методів хімічного аналізу. 2. Нові можливості фізико-хімічних методів аналізу. 3. Принципи класифікації ФХМА. 4. Методичні засоби ФХМА. 5. Етапи хімічного аналізу об'єктів. Їх призначення. 6. Характеристика аналітичного сигналу у ФХМА. 7. Які параметри аналітичного сигналу можуть використовуватися для якісного, а які для кількісного аналізу? 8. Що таке калібрування? З допомогою чого воно проводиться? 9. Вимоги до комплекту еталонів (стандартів). 10. Які бувають способи калібрування? 11. Коли використовуються способи прямого (абсолютного) калібрування? 12. Коли необхідно використати способи відносного калібрування?
Питання для самоконтролю. 1. На чому грунтуються молекулярно-абсорбційні методи аналізу. 2. Структура енегетичних рівнів молекул. 3. Аналітичний сигнал молекулярно-абсорбційного аналізу 4. Чому спектр поглинання молекул має смугастий характер? 5. Що таке характеристичні або групові смуги? Для чого вони використовуються? 6. Закон Бугера-Ламберта-Бера. Коефіцієнт поглинання, екстинкція. 7. Причини відхилень від закону Бера. 8. Схема приладів для вимірювання спектра поглинання. 9. Призначення фотоколориметрів, фотоелектроколориметрів. 10. Вимоги до забарвлених сполук і реакцій їх утворення. 11. Етапи розробки фотоколориметричних методик аналізу. 12. Способи проведення кількісного фотоколориметричного аналізу. 13. Визначення декількох речовин, спектри яких перекриваються. 14. Переваги методу диференційної фотометрії. 15. Фотоелектроколориметричне титрування. 16. Основи та можливості турбідиметрії. 17. Основи та можливості нефелометрії.
Способи детектування
В детектор поступають бінарні суміші компонентів проби з газом носієм, тому принцип детектування може грунтуватися на вимірюванні будь-якої фізичної властивості компонентів, яка відмінна від властивостей газу-носія, або специфічних властивостей речовин. У газовій хроматографії використовуються такі принципи детектування: 1. Залежність теплопровідності газової суміші від її складу (детектор за теплопровідністю). 2. Тепловий ефект спалювання горючих компонентів (термохімічний детектор). 3. Іонізація органічних сполук у полум'ї водню (полум'яно-іонізаційний детектор). 4. Іонізація органічних сполук під дією радіоактивного випромінювання (аргоновий детектор). 5. Захоплення електронів молекулами органічних сполук (детектор за захопленням електронів). 6. Зменшення іонізації полум'я з атомами лужних металів у присутності фосфор- і галогено-похідних (термоіонний детектор). 7. Залежність густини газової суміші від її складу (детектор за густиною). 8. Специфічне випромінювання фосфор- і сірковмісних сполук у полум'ї водню (полум'яно-фотометричний детектор). 9. Залежність перепаду тиску на діафрагмі від складу газової суміші (діафрагмовий детектор). Крім цих методів, постійно розробляються нові принципи детектування для забезпечення більшої чутливості аналізу. Найбільш розповсюдженими детекторами є детектори за теплопровідністю і полум'яно-іонізаційні детектори.
3.7.1. Детектор за теплопровідністю (ДТП). Принцип дії детектора грунтується на залежності теплопровідності газової суміші від її складу. Теплопровідність суміші газів є адитивною функцією і сигнал детектора залежить від різниці теплопровідностей газу-носія і компонента суміші. Для збільшення чутливості як гази-носії використовують He або H2, які мають найбільшу теплопровідність. Теплопровідність газової суміші вимірюється шляхом вимірювання опору провідника (W, Pt), вміщеного в потік елюату. Зміна теплопровідності газу навколо провідника призводить до порушення теплового балансу і зміни температури провідника, яка впливає на його опір. Опір провідника, пропорційний вмісту компонента в елюаті, вимірюється мостовою схемою і фіксується за допомогою самописця. Цей детектор відрізняється простотою конструкції, надійністю і має достатню чутливість (10-3 % об. за пропаном при використанні Не як газу-носія). Недоліком детектора є можливість корозії матеріалу провідника при підвищених температурах.
3.7.2. Полум'яно-іонізаційний детектор (ПІД). На виході з хроматографічної колонки до елюенту домішується постійний потік водню. Суміш виходить крізь пальник і згоряє в атмосфері повітря між двома металічними електродами. До електродів прикладається постійна напруга 40-300 В. Якщо з колонки виходить тільки газ носій (Ar, N2), полум'я слабко іонізоване і між електродами проходить струм порядка 10-14 А. При попаданні з колонки в полум'я органічних сполук, вони іонізуються, внаслідок чого між електродами проходить струм іонізації, сила якого досягає 10-10 - 10-8 А. Іонізаційний струм пропорційний кількості заряджених частинок, а значить і концентрації органічних сполук. Висока чутливість ПІД (10-4 - 10-5 %), зробили його найрозповсюдженішим детектором для визначення мікродомішок і для роботи з капілярними колонками. Чутливість ПІД пропорційна кількості атомів вуглецю в молекулі вуглеводню. Недоліком ПІД є нечутливість його до неорганічних сполук, таких як оксиди вуглецю, сірки, фосфору, H2O, і деяких органічних сполук - CH2O, HCOOH.
Кількісний аналіз
Кількісним параметром хроматографічного піка є його площа (S), яка пропорційна масі речовини введеної в хроматограф з пробою (3.20). З теорій хроматографії видно, що при збільшенні кількості речовини збільшується висота піка (h) при незмінній його ширині (ω 0,5). Тому висота піка теж є кількісним параметром піків. Висоту піків виміряти просто. Для вимірювання площі піків необхідно застосовувати спеціальні засоби або реєстратори з інтеграторами, але залежність площі піків від маси речови має більший діапазон лінійності, ніж висота. Тому як кількісні характеристики використовують також величини, пропорційні площі піків - hω0,5 або hlr. Таким чином, кількісним параметром (Р) можуть бути як площа піка (S), так і його висота (h) або добуток висоти на ширину піка (hω0,5) чи на відстань затримування (hlr). Тоді для і -го компонента можна записати: mi = kiּPi. (3.26) З другого боку, mi = CiVпрMi, (3.27) де Ci, Mi - молярна концентрація та молярна маса компонента, Vпр - об'єм проби, введеної в хроматограф. З рівнянь 3.26 та 3.27 одержимо:
Величина ki залежить від властивостей речовини, конструкції і режиму роботи хроматографа, тому методом абсолютного калібрування можна користуватися при можливості підтримування стабільного режиму роботи хроматографа і введення однакового об'єму проби та калібрувальних сумішей. Газові хроматографи зазвичай комплектуються дозаторами газових проб, які дають можливість вводити постійний об'єм (1 - 10 мл) проби газових сумішей, і користуватися методом абсолютного калібрування або калібрувальних коефіцієнтів. Рідкі проби об'ємом 1 - 10 мкл вводяться мікрошприцами, які не забезпечують постійності об'єму проб і метод абсолютного калібрування для точного аналізу рідин не використовується. У цьому випадку користуються методом відносного калібрування (внутрішнього стандарта). Він полягає у тому, що до відомої маси проби (mпр) додають відому масу речовини стандарта, якої немає в пробі (mст), змішують їх і хроматографують одержану суміш. Масові частки компонентів проби розраховують за формулою:
де Pi, Pст - кількісні параметри піків і -го компонента та речовини стандарта, fi - нормувальний коефіцієнт, який дорівнює відношенню чутливостей хроматографа до компонента і речовини стандарта. Нормувальний коефіцієнт розраховують з хроматограми суміші, яка містить відомі концентрації компонентів (wi) та речовини стандарта (wст, % мас):
У випадку, коли необхідно визначати концентрації всіх компонентів суміші, суму їх концентрацій приймають за 100% і тоді відпадає необхідність в додаванні речовини стандарта. В цьому випадку розрахунки концентрацій проводять за методом внутрішньої нормалізації:
Нормувальні коефіцієнти розраховують за формулою 3.30, прийнявши одну з речовин за стандартну, при цьому fст = 1.
Питання для самоконтролю. 1. На чому грунтуються хроматографічні методи аналізу? 2. Суть хроматографічного розділення. 3. Класифікація хроматографічних методів аналізу. 4. Принципова схема і призначення основних блоків газового хроматографа. 5. Теорія рівноважної газової хроматографії. 6. Теорія нерівноважної газової хроматографії. 7. Теорія тарілок. Ефективність хроматографічної колонки. 8. Рівняння Ван-Деемтера. 9. Хроматограма та її характеристики. Аналітичний сигнал хроматографічних методів аналізу. 10. Якісні та кількісні характеристики аналітичного сигналу. 11. Чіткість хроматографічного розділення компонентів. 12. Вплив основних факторів на чіткість розділення. 13. Вимоги до рідкої нерухомої фази, носіїв рідкої фази і твердої нерухомої фази. 14. Способи детектування. Принцип дії детекторів за теплопровідністю та іонізацією полум’я. 15. Можливості якісного хроматографічного аналізу. 16. Кількісні параметри хроматографічного піка. 17. Кількісний аналіз газових сумішей. 18. Методи внутрішнього стандарта, внутрішньої нормалізації. 19. Застосування хроматографічних методів аналізу.
Питання для самоконтролю. 1. Класифікація електрохімічних методів аналізу. 2. Основи потенціометричних методів аналізу. 3. Класифікація електродів. Електроди I, II, III роду, індиферентні та іонообмінні. 4. Аналітичний сигнал потенціометричних методів аналізу. 5. Вимоги до електродів. 6. Індикаторні та стандартні електроди. 7. Вимірювання потенціалів в потенціометрії. 8. Пряма потенціометрія. Переваги. 9. Потенціометричне титрування, його можливості. 10. Потенціометричне титрування, основане на реакціях нейтралізації, окиснення-відновлення, осадження, комплексонометрії.
Питання для самоконтролю. 11. Основи полярографічних методів аналізу. 12. Види поляризації. 13. Принципова схема полярографічної установки. 14. Переваги та недоліки ртутного краплинного електрода. 15. Полярографічна хвиля. Граничний дифузійний струм. 16. Якісний полярографічний аналіз. 17. Способи проведення кількісного полярографічного аналізу. 18. Причини спотворення форми полярограм, полярографічні максимуми і їх подолання. 19. Амперометричне титрування, його можливості. 20. Способи збільшення чутливості полярографічних методів аналізу.
Питання для самоконтролю. 20. На чому грунтуються кондуктометричні методи аналізу? 21. Електрична провідність розчинів електролітів – питома, еквівалентна, рухливість. 22. Принципова схема кондуктометричної установки. 23. Прямий кондуктометричний аналіз, його переваги і недоліки. 24. Кондуктометричне титрування, його можливості. 25. Закон Фарадея. 26. Вихід за струмом. 27. Потенціостатична кулонометрія. 28. Амперостатична кулонометрія. 29. Кулонометричне титрування.
Питання для самоконтролю. 30. Загальна характеристика радіохімічних методів аналізу. Поняття про природні та штучні радіоізотопи (радіонукліди). 31. Типи ядерних перетворень (розпадів). Характеристика видів радіоактивного випромінювання. 32. α-розпад та характеристика α-випромінювання. 33. β–-розпад (електронний розпад) та характеристика β–-випромінювання. 34. β+-розпад (позитронний розпад. 35. Ізомерний перехід (ІП) та характеристика γ-випромінювання. 36. Спонтанний поділ (f). 37. Закон радіоактивного розпаду. Активність (абсолютна, відносна) та одиниці активності. 38. Зв’язок активності з масою радіонукліду. 39. Методи реєстрації радіоактивного випромінювання. 40. Якісний радіометричний аналіз. 41. Кількісний радіометричний аналіз. 42. Способи, основані на вимірюванні природної радіоактивності. 43. Способи, основані на вимірюванні штучної радіоактивності. 44. Переваги та недоліки радіометричних методів аналізу.
[PS1]була добутком або сумою двох функцій!!!! [PS2]Що це таке і де воно на рисунку? [PS3]Забрати [PS4]Не зрозумів [PS5]В попередньому розділі нумерація формул відсутня, а тут знову починається. Привести все до одного виду!!!!!!!!!! [PPS6]Визначитись з рисунками. Васильев, с190 [PS7]Ляликов, с. 367, р. Х-2
Особливості фізико-хімічних (інструментальних) Методів аналізу
Основне завдання аналітичної хімії - встановлення якісного та кількісного складу речовин, а також ідентифікація та встановлення будови молекул нових речовин. Класичні методи аналітичної хімії (хімічні методи аналізу) - гравіметричний та титриметричний аналіз - дозволяють визначати якісний компонентний склад речовин з межею виявлення 10-3 - 10-5 %. Діапазон кількісного визначення концентрацій компонентів складає 0,01 – 100 % при відносній точності результатів аналізу 0,2 %. Хімічні методи аналізу характеризуються використанням простого обладнання але вимагають застосування великої кількості ручних операцій і тривають довгий час (від десятків хвилин до декількох годин), слабко піддаються автоматизації. Кількісний аналіз речовин, вміст яких не перевищує 0,01 %, практично неможливий. Такі показники хімічних методів аналізу не задовольняють запити сучасної хімічної промисловості та промисловості виробництва матеріалів для будівництва, сільського господарства, радіоелектроніки, космічної техніки, атомної енергетики, медицини, наукових досліджень. Перед аналітичною хімією стоїть завдання розробки методів аналізу з межею виявлення 10-5 - 10-10 % і нижче в процесах виробництва надчистих матеріалів. При аналізі об'єктів навколишнього середовища і екологічному контролі діючих виробництв необхідно проведення аналізу великої кількості проб повітря, стічних вод, відходів виробництва. Це вимагає розробки експресних автоматизованих методик аналізу. Інколи виникає необхідність аналізувати об'єкт без руйнування (без відбирання проби) або визначати не середній склад, а склад в деяких точках на поверхні чи в об'ємі об'єкта (локальний аналіз). Цим вимогам найбільш повно відповідають фізико-хімічні методи аналізу (ФХМА). Відмінність ФХМА від хімічних методів полягає в тому, що для одержання видимого аналітичного сигналу використовують прилади, які перетворюють яку-небудь властивість хімічної системи в переважно електричний сигнал, який легко зареєструвати вимірювальними приладами або записати у вигляді графіків на паперових носіях чи дисплеях. Фізико-хімічні методи аналізу поділяються на 2 групи: 1. Власне фізико-хімічні методи, які грунтуються на вимірюванні фізичних або фізико-хімічних властивостей (параметрів) системи при проведенні хімічної реакції з об'єктом аналізу. 2. Фізичні методи аналізу, які грунтуються на вимірюванні фізичних властивостей (параметрів) системи без проведення хімічних реакцій. Між цими групами методів чіткої границі немає, об'єднує їх те, що аналітичний сигнал вимірюється за допомогою приладів (інструментів), тому інколи ці методи називаються інструментальними. Класифікація ФХМА основана на спільності теоретичних і практичних принципів одержання аналітичного сигналу. Загальне число ФХМА перевищує декілька десятків, але найбільш поширені такі: 1. хроматографічні методи аналізу; 2. спектральні (оптичні) методи; 3. електрохімічні методи; 4. радіометричні методи; 5. масспектрометричні методи; 6. рентгеноспектральні методи. Крім методів, в яких використовуються прилади, що грунтуються на одному принципі, існують методи, в яких використовуються різні принципи для одержання аналітичного сигналу. Такі методи називаються гібридними. Майже у всіх ФХМА для встановлення складу речовини використовується 2 методичних засоби: метод прямих вимірювань і метод титрування. У загальному вигляді, процес одержання даних про хімічний склад об'єкта аналізу в прямих методах складається з таких етапів (рис. 1.1):
Рис. 1.1. Загальна схема проведення аналізу
|
|||||||||
|
|
Последнее изменение этой страницы: 2017-02-07; просмотров: 277; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.144.84.155 (0.209 с.) |

 (1.1)
(1.1)
 (1.3)
(1.3)
 (1.4) або
(1.4) або
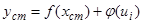 (1.5)
(1.5) (1.6)
(1.6) (1.7)
(1.7)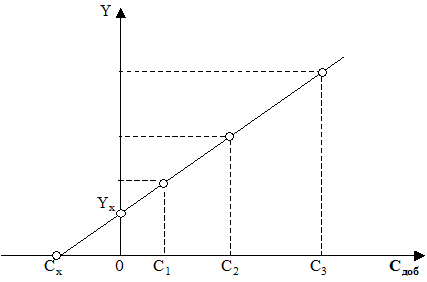
 . (1.8)
. (1.8) . (1.9)
. (1.9) . (1.10)
. (1.10) , (1.11),
, (1.11), (1.12)
(1.12) . (3.28)
. (3.28) , (3.29)
, (3.29)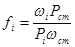 . (3.30)
. (3.30) . (3.31)
. (3.31)


