
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Розподіл кальцію в організмі
Загальна кількість кальцію в тілі дорослої людини досягає 1 кг, близько 99 % якого локалізовано в кістках, де кальцій разом із фосфатами утворює кристали гідроксіапатиту, що складають основу неорганічної структури скелета. Гомеостаз кальцію Як загальна кількість кальцію в організмі, так і концентрація його іонізованої форми в екстрацелюлярних просторах та всередині клітин, тобто гомеостаз кальцію, визначається функціонуванням таких анатомо-фізіологічних систем: 1. Кісток скелета — резервуара кальцію. Клітинні елементи кісткової тканини здійснюють не тільки утворення кісток, але й виконують найважливішу функцію контролю кальцієвого гомеостазу в організмі: а) завдяки діяльності клітин, що утворюють кісткову тканину (остеобластів) відбувається як біосинтез компонентів остеоїду — органічного матриксу кісткової тканини (містить колаген I типу — близько 90 % всіх білків кісткового матриксу, глікопротеїни та протеоглікани), так і відкладення впродовж колагенових фібрил кристалів гідроксіапатиту кальцію, тобто мінералізація остеоїду; б) завдяки функціонуванню остеокластів (похідних моноцитів) — клітин, що здійснюють резорбцію (resorptio — лат.) кісткової тканини, відбувається звільнен- ня кальцію, що зв’язаний з органічним матриксом кісток, та вихід Са2+ в кров. 2. Тонкої кишки, у верхніх відділах якої здійснюється всмоктування (абсорбція та реабсорбція) кальцію і фосфатів, які споживаються у складі продуктів харчування або надходять у порожнину кишечника внаслідок звільнення цих іонів у процесі метаболізму. 3. Нирок, уздовж канальців яких відбувається реабсорбція іонів кальцію та У свою чергу, ефекторна функція кісток, кишеч- ника та нирок відносно обміну та гомеостазу кальцію є об’єктом гуморального контролю з боку трьох фізіо- логічно активних сполук, які є головними регуля- торами кальцієвого балансу в організмі: паратгормону, кальцитріолу (вітаміну D 3) та кальцитоніну (рис. 25.4). Паратгормон Паратгормон (паратиреоїдний гормон) — спо- лука, що синтезується в головних і ацидофільних клітинах паращитовидних залоз.За хімічною природою є простим білком (м.м.= 9,5 кД), який має один поліпептидний ланцюг, що складається з 84 амінокислотних залишків. Паратгормон синтезується на рибосомах у формі препропаратгормону (115 амінокислотних залишків), який підлягає процесингу в ендоплазматичному ретикулумі та апараті Гольджі з утворенням спочатку пропаратгормону (90 амінокислотних залишків), а потім — паратгормону. Паратгормон має гіперкальціємічний ефект, водночас зменшуючи концентрацію в крові фосфатів, що є результатом його впливу на обмін цих сполук в таких ефекторних системах: Кальцитріол — сполука гормонального типу дії, що утворюється в організмі з біологічного попередника, яким в організмі людини та вищих тварин є жиророзчинним вітаміном D3 (холекальциферол).
. Порушення кальцієвого гомеостазу Найбільш поширеними клінічно окресленими порушеннями гомеостазу кальцію є патологічні синдроми, пов’язані з дефіцитом вітаміну D3 (які проявляються як рахіт у дітей і різні форми остеопорозу в дорослому та похилому віці) та захворювання, спричинені первинною патологією паращитовидних залоз — гіпер- та гіпопаратиреоз. Рахіт — захворювання дитячого віку, яке спричиняється зменшеним надходженням та (або) синтезом в організмі вітамінів групи D — тваринного D3 (холе- кальциферолу) та рослинного D2 (ергокальциферолу). Вірогідність захворювання значно зростає в умовах недостатнього опромінення шкіри дитини сонячним світлом, що є необхідним для утворення вітаміну D3 з 7 дегідрохолестерину. Основними проявами рахіту є гіпокальціємія та гіпофосфатемія, які призводять до глибоких порушень кальцифікації кісткової тканини та специфічних змін скелета. Гіперпаратиреоз — група захворювань, розвиток яких пов’язаний із надлишковою секрецією паратгормону, аномальним збільшенням внаслідок цього концентрації кальцію в сироватці крові і гіпофосфатемією. Первинний гіперпаратиреоз — патологія, що спричиняється наявністю в паращитовидних залозах гормонально активних пухлин — аденоми, карциноми або гіперплазією залози. Провідними симптомами захворювань цієї групи є ураження кісткової системи (проявляється демінералізацією кісток — остеопорозом) та нирок із розвитком сечокам’яної хвороби (внаслідок відкладання солей та утворення __
58.МеханІЗМИ ПЕРЕТВОРЕННЯ ПОЖИВНИХРЕЧОВИН У ТРАВНОМУ ТРАКТІ Перетравлення поживних речовин (білків, вуглеводів, ліпідів) — це процесс гідролізу відповідних сполук у складі продуктів харчування, що відбувається в травному каналі і призводить до утворення простих біомолекул, які за рахунок дії спеціальних механізмів мембранного транспорту всмоктуються у кров. Початкові процеси травлення відбуваються в ротовій порожнині за участю слини, яка є біологічною рідиною з рН 6,8, що на 99,5 % складається з води і містить різноманітні білки (ферменти, муцини, імуноглобуліни, лізоцим тощо) та неорга- нічні солі. Слина має змащувальну дію відносно сухих продуктів харчування, під її впливом полегшується процес жування та створюються умови для подальшого перетворення компонентів харчування під впливом специфічних ферментів. До ферментів слини належать глікозидази, що каталізують певні процеси гідролізу вуглеводів — α-амілаза та мальтаза. Під дією цих ферментів можливе розщеплення крохмалю до високомолекулярних декстринів і мальтози до глюкози,але, оскільки час впливу слини на харчову грудку незначний, — відповідніпродукти утворюються в порожнині рота лише в незначній кількості.Основні процеси травлення поживних речовин їжі відбуваються в шлунку —розщеплення білків до пептидних молекул та в різних відділах тонкого кишечника — розщеплення пептидів, вуглеводів, жирів (ліпідів). Шлунок виробляє власні протеолітичні ферменти (пепсиноген, ренін); кишечник (залози Брунерата Ліберкюна) синтезує деякі пептидази, дисахаридази, фосфоліпази та полінуклеотидази. Травлення в кишечнику неможливе без участі гідролітичних ферментів, які надходять сюди з підшлункової залози — протеаз (трипсину, хімотрипсину,Продукти харчування Білки Вуглеводи Ліпіди (жири тощо)еластази), карбоксипептидази, амілаз, ліпаз. У процесі травлення жирів берутьучасть біохімічні компоненти жовчі, що синтезуються в гепатоцитах печінки. Перетравлення білків Біохімічні процеси перетравлення білків та пептидів, що надходять до організму людини з їжею, відбуваються в шлунку й тонкій кишці. Гідроліз цих компонентів відбувається під дією ферментів, які виробляються клітинами слизової оболонки травного каналу й екзокринної частини підшлункової залози. Протеази шлунка,кишечника та підшлункової залози гідролізують певні пептидні зв’язки в молекулах білків і пептидів їжі, і в результаті їх послідовної дії утворюється суміш вільних L-амінокислот та найпростіших пептидів, що транспортуються всередину ентероцитів і далі — в кров’яне русло. Перетравлення білків у порожнині шлунка Шлунковий сік, під дією якого відбувається гідроліз білків, — це кисла рідина з рН 1,5-2,5. Основними біохімічними компонентами шлункового соку, що беруть участь вперетворенні білків продуктів харчування, є соляна кислота та протеолітичний фермент пепсин. Крім того, до складу шлункового соку входять кислі фосфати (переважноNaH2PO4) та деякі органічні кислоти, складаючи загальну кислотність шлунка.Соляна кислота виробляється вспеціальних обкладинних (оксинтних)клітинах слизової оболонки шлунка за участю хлоридів, які надходять із крові.Донором протонів, необхідних для утворення HCl, є вугільна кислота, що утворюється з Н2О та СО2 за участю карбоангідрази:Секреція іонів Н+ в порожнину шлунка відбувається при дії протонної помпи мембран оксинтних клітин — Н+, К+-АТФази (рис. 26.2). Концентрація HCl у шлунковому соку складає 0,45-0,60 %. Соляна кислота необхідна для утворення активного ферменту пепсину і прояву максимуму його каталітичної активності.
Пепсин — протеаза з м.м. 42 кД, що синтезується головними клітинами слизової оболонки шлунка у вигляді проферменту пепсиногену (м.м. 35 кД). Початкові етапи перетворення пепсиногену в пепсин здійснюються за участю іонів Н+, які сприяютьвідщепленню від молекули проферменту N-кінцевого захисного пептиду, що супроводжується розкриттям активного центру; в подальшому процес стає автокаталітичним — молекули пепсину спричиняють власне утворення з проферменту:За механізмом дії пепсин є ендопептидазою, що специфічно атакує пептидні зв’язки, в утворенні яких беруть участь залишки ароматичних (фенілаланіну, тирозину), а також дикарбонових (глутамату, аспартату) амінокислот. Під дією пепсину білки розшеплюються на великі поліпептидні фрагменти — пептони, гідроліз яких завершується в тонкій кишці.Реннін (хімозин, сичужний фермент) — протеаза, що міститься в шлунковому соку новонароджених дітей. Реннін є ферментом, який за участю іонів Са2+ спричиняє перетворення розчинних білків молока — казеїнів у нерозчинні — параказеїни, які підлягають протеолітичній дії пепсину (“створожування молока”).Перетравлення білків в кишечнику Частково перетравлена напіврідка маса поживних сполук, що утворюється в шлунку (хімус) періодично надходить через пілоричний клапан у дванадцятипалу кишку. В цю ж частину травного каналу надходять із підшлункової залози протеолітичні ферменти та пептидази, які діють на пептиди, що надходять зі шлунка. Ката літична дія цих ферментів відбувається в слабколужному середовищі (рН 7,5-8,0), яке утворюється наявними в кишковому соку бікарбонатами NaHCO3. Більшість ферментів протеолітичної дії, що функціонують у тонкій кишці, синтезуються в екзокринних клітинах підшлункової залози у вигляді проферментів, які активуються після їх надходження в дванадцятипалу кишку (трипсиноген, хімотрипсиноген, проеластаза, прокарбоксипептидази А і В). Гідроліз білків та пептидів, що надходять із шлунка, відбувається як у порожнині тонкої кишки, так і на поверхні ентероцитів — пристінкове, або мембранне травлення. Трипсин — протеолітичний фермент з м.м. 24,7 кД, що утворюється в порожнині кишечника з неактивного проферменту трипсиногену під дією ентерокінази, яка відщеплює від молекули проферменту N-кінцевий гексапептид з утворенням каталітично активного трипсину: Трипсин є ендопептидазою, яка найбільш активна відносно пептидних зв’язків, утворених основними амінокислотами аргініном та лізином. Хімотрипсин — протеолітичний фермент (м.м. 29 кД), який утворюється з проферменту хімотрипсиногену за каталітичної дії трипсину, що відщеплює від молекули проферменту декілька інгібіторних пептидів:Хімотрипсин є ендопептидазою, яка розщеплює до 50 % пептидних зв’язків вмолекулах білків та пептидів їжі, в тому числі зв’язків, нечутливих до дії пепсину та трипсину.Еластаза — ендопептидаза, що також має широку субстратну специфічність,розщеплюючи пептидні за’язки, що утворюються залишками амінокислот малого розміру — гліцину, аланіну, серину. Утворені при дії зазначених вище ендопептидаз короткі пептиди підлягають дії екзопептидаз кишечника — карбоксипептидаз А і В, амінопептидаз та дипептидаз. Карбоксипептидази — пептидази, що гідролізують пептидні зв’язки, утворені С-кінцевими амінокислотами: карбоксипептидаза А відщеплює від С-кінця амінокислоти з гідрофобними радикалами, а карбоксипептидаза В — С-кінцеві залишки лізину й аргініну. Амінопептидази — ферменти ентероцитів, що відщеплюють від коротких пеп- тидів N-кінцеві амінокислотні залишки.
Дипептидази — пептидогідролази, що розщеплюють дипептиди до вільних амінокислот. Послідовна дія всього набору шлункових, панкреатичних і кишечних пептидогідролаз забезпечує повне розщеплення білків та пептидів продуктів харчування до амінокислот. У кровотік слизовою оболонкою кишечника всмоктуються тільки вільні амінокислоти. Перетравлення вуглеводів Основні реакції розщеплення вуглеводів відбуваються в тонкому кишечнику за рахунок дії ферментів підшлункової залози, що потрапляють у порожнину дванадцятипалої кишки, і власних ферментів кишкового соку. Подібно до перетворення білків та пептидів, поряд з порожнинним травленням, у кишечнику відбувається пристінкове (на поверхні мембран ентероцитів) травлення вуглеводів. Амілази, що діють у кишечнику — це ферменти α-амілаза (переважно) та β-амілаза, які синтезуються в підшлунковій залозі. Панкреатична α-амілаза — це ендоглікозидаза, подібна до ферменту слини, яка гідролізує крохмаль та глікоген з утворенням суміші розгалужених і нерозгалужених олігосахаридів і деякої кількості мальтози і мальтотріози. β-Амілаза — панкреатична екзоглікозидаза, яка відщеплює від нерозгалужених гомополісахаридних ланцюгів залишки мальтози Дисахаридази та олігосахаридази — ферменти, що синтезуються в тонкій кишці і спричиняють розщеплення до моносахаридів відповідних цукрів, які утворюються як продукти дії амілаз або надходять до травного каналу в складі рослинних продуктів харчування: мальтаза (α-глюкозидаза) — фермент, що гідролізує мальтозу та відщеплює термінальні глюкозні залишки з нередукуючих кінців α(1 4)-зв’язаних олігосахаридів; мальтаза та ізомальтаза (α(1 6)-глікозидаза) завершують розщеплення гомополісахаридів, розпочате амілазами; лактаза (β-галактозидаза) — фермент, що розщеплює лактозу (молочний цукор) до двох моносахаридів — галактози та глюкози; надзвичайно велике фізіологічне значення лактази в харчуванні дітей; сахараза (β-фруктозидаза) — фермент кишечного соку, що гідролізує з утворенням глюкози і фруктози дисахарид сахарозу — основний компонент бурякового та тростинного цукру. Внаслідок дії зазначених глікозидазних ферментів на рослинні та тваринн вуглеводи продуктів харчування утворюється суміш моносахаридів (в основному глюкози, фруктози й галактози), які всмоктуються клітинами кишкового епітелію і поступають у кров. Глюкоза складає до 90 % усіх моносахаридів крові, решту становлять інші гексози та пентози, утворюючи в сумі загальний цукор крові (4,5-6,5 ммоль/л). Недостатність дисахаридаз Існує група спадкових ензимопатій, що пов’язані з недостатністю синтезу і виділення в кишковий сік ферментів, які гідролізують дисахариди. Ці ферментні дефекти проявляються порушеннями у перетравленні та всмоктуванні відповідних цукрів. Недостатність лактази Спадковий дефіцит ферменту призводить до неспроможності кишкового соку розщеплювати молочний цукор, і позначається як непереносимість лактози. У відносно рідкісних випадках патологія (спадкова відсутність лактази) в умовах харчування материнським молоком клінічно проявляється вже в перші дні життя новонародженого. Проте, в більшості випадків ензимопатія зстрічається у вигляді низької активності лактази. Ця форма непереносимості лактози успадковується як автосомна рецесивна патологія і вперше проявляється в підлітковому періоді або у молодому віці. Розповсюдженість недостатності лактази широко варіює в різних расових групах, особливо часто зустрічаючись у жителів східних країн та кольорового населення Північної Америки. Так, зокрема частота патології у датчан становить 3 %, а в тайців — 97 %.
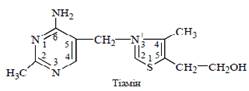
кількості нейтральних жирів їжі; вони можуть всмоктуватися епітелієм слизової оболонки тонкої кишки в негідролізованій формі \і розщеплюються до гліцерину та жирних кислот всередині ентероцитів. Всмоктування продуктів перетравлення ліпідів Складна суміш продуктів гідролізу ліпідів, зазначена вище, утворює ліпідні міцели, які можуть абсорбуватися слизовою оболонкою кишечника, і практично всі жири харчових продуктів надходять до грудної лімфатичної протоки завдяки всмоктуванню цих міцел. Проникнення ліпідних міцел всередину ентероцитів відбувається шляхом піноцитозу або дифузії окремих ліпідних молекул через апікальну мембрану клітин. Порушення процесів перетравлення ліпідів Порушення гідролізу та всмоктування харчових ліпідів у кишечнику супроводжуються розвитком стеатореї — наявності збільшеної кількості жирів у фекальних масах. Розрізняють такі види порушень перетравлення ліпідів у кишечнику людини (А.Ш.Бышевский, О.А.Терсенов, 1994): 1) дефіцит панкреатичної ліпази, що спричинений захворюваннями підшлункової залози — панкреатична стеаторея; 2) дефіцит жовчі в кишечнику, пов’язаний з захворюваннями печінки або жовч- них шляхів — гепатогенна стеаторея; 3) пригнічення ферментних систем ліполізу та ресинтезу триацилгліцеролів у кишечнику при його захворюваннях — ентерогенна стеаторея. 59 Поруш травлення нутрыэнтыв у шлунку та кишечнику Былки 1-1,5 г/кг за добу Квашиа ркор (у дытей),алыментарний маразм (у дорослих) Клынычны прояви 1 набряки (зменшнння кылькосты альбумыныв)2_Жирова ынфыльтрацыя печынки(знижена конц ЛПДНЩ) 3_Анемыя (виникаэ апатія)4_В ШКТ знижується синтез ферментів,які відповіають за перетравлення(трипсин.лактоза) Вугливоди –ензимопатії пов’язані з недостатністю ферментів які гідролізують дисахариди і виникають порушення у перетравленні та всмоктуванні відповідних цукрі (лактази,сахарази) Ліпіди ензимопатії характериз стеатореєю (підвищення кількості жирів у фекаліях)у кишечнику є такі порушення перетравл ліпідів: 1_Панкреатична стеотолрея (дефіцит панкреатичної ліпази)2_Гепалтогенна стеаторкя (дефіцит жовчі у кишечнику)3_ентерогенна стеаторея (поруш лі полізу триацигліцеролів) 60 Мікроелементи в харчув люд вітаміни Мікроелементи; вітаміни мінеральні реч,вода Вітам.не синтез. В організмі люди.Це незамінні компоненти їжі.Потреба від декількох мкг до десятків мкг. Водорозчинні: В1 (тіамін), _В2(рибофлавін), РР(В5,або ніацин),В6(піридоксин),В12(кобаломін),фолієва кислота,Н(біотин),пантенонова кислота,С,Р Жиророзчинні: А (ретинол),К,Е,F,Д(кальциферол) Мінеральні реч джерелом є біологічні компоненти їжі.Вода-незамінний компонент 1750-2200г.Вітамінна недостатність –гіповітаміназ,авітаміноз.Супроводж важкими розладами біохім і фізіол процесів. Причіни:1_зменшення або відсутність надходження віт в організм у складі продуктів харчуванняю2Поруш засвоєння вітамінів 3_підвищене виведення вітам з організму Вітамін В1 Вітамін В1 (тіамін) за хімічною будовою є продуктом конденсаціїдвох гетероциклічних сполук —похідного піримідину (2-метил-5-гідроксиметил-6-амінопіримідину) та тіазолу (4-метил-5-гідроксіетилтіазолу):Біологічні властивості та механізм діїБіологічна активність вітаміну В1 полягає в участі в енергетичному, зокрема вуглеводному, обміні. Біохімічній механізм дії вітаміну зумовлений його коферментноюформою — тіаміндифосфатом (ТДФ) (“кокарбоксилазою”), який утворюється в результаті фосфорилювання вільного тіаміну за участю ферменту тіамінфосфокінази: тіамін + АТФ тіаміндифосфат + АМФ. Коферментні функції ТДФ пов’язані з каталізом таких біохімічних реакцій: – окислювальне декарбоксилювання пірувату (як компонент мультиензимного піруватдегідрогеназного комплексу); – окислювальне декарбоксилювання α-кетоглутарату в циклі Кребса (як компонент α-кетоглутаратдегідрогеназного комплексу); – транскетолазні реакції пентозофосфатного шляху окислення глюкози (як кофермент транскетолази). Згідно із вищерозглянутою роллю в перебігу ключових реакцій аеробного обміну вуглеводів, характерним біохімічним проявом недостатності вітаміну В1 є накопичення в крові та внутрішніх тканинах надлишкового (неокисленого) пірувату. Клінічно недостатність тіаміну проявляється розвитком уражень периферичної нервової системи (поліневропатії, поліневриту аж до розвитку атрофічного паралічу кінцівок), виражених порушень з боку міокарда, секреторної та моторної функцій шлунка. Класична форма авітамінозу В1 — захворювання бери-бері — спостерігалася у населення Південно-Східної Азії в умовах тривалого харчування полірованим рисом, позбавленим тіаміну, що міститься в зовнішній оболонці зернини. Джерела та добова потреба Основними джерелами вітаміну В1 у харчуванні людини є продукти рослинного походження, зокрема хліб (переважно житній, з борошна грубого помолу), крупи (гречана, вівсяна); багаті на тіамін (та інші вітаміни групи В) пивні дріжджі. Добова потреба в тіаміні складає 1,5-2,0 мг* [1,5 мг]**.
Вітамін В2 За хімічною будовою вітамін В2 (рибофлавін) є похідним трициклічної сполуки ізоалоксазину та спирту рибітолу —6,7-диметил-9-рибітилізоалоксазин: Біологічні властивості та механізм дії Біологічні функції вітаміну В2 полягають у його участі в окислювально-відновлювальних реакціях. Коферментними формами рибофлавіну є ФАД (флавінаденіндинуклеотид) та ФМН (флавінмононуклеотид) — простетичні групи багатьох анаеробних та аеробних дегідрогеназ та оксидаз, що беруть участь в окисленні численних інтермедіатів вуглеводного, ліпідного та амінокислотного обміну. Джерела та добова потреба Рибофлавін міститься в багатьох продуктах рослинного та тваринного походження, тому в умовах звичайного мішаного харчування людина отримує необхідну для нормальної життєдіяльності кількість вітаміну. Добова потреба в рибофлавіні складає 2,0-2,5 мг* [1,7 мг]**.
Вітамін РР (вітамін В5) Властивості вітаміну РР (ніацину) мають нікотинова кислота (піридин-3-карбонова кислота) та її амід, які є в організмі взаємно перетворюваними молекулярними формами: Біологічні властивості та механізм дії Вітамін РР є необхідним фактором для перебігу багатьох біохімічних реакцій, пов’язаних із окисленням субстратів вуглеводного, ліпідного, амінокислотного та інших видів метаболізму. Коферментними формами вітаміну РР є коферменти анаеробних дегідрогеназ НАД (нікотинамідаденіндинуклеотид) та НАДФ (нікотинамідаденіндинуклеотидфосфат), до складу яких входить амід нікотинової кислоти. Недостатність вітаміну РР проявляється характерними патологічними змінами шкіри, особливо вираженими в умовах її сонячного опромінення. Дерматит, специфічний для цього гіпо- (а-) вітамінозу, отримав назву “пелагри” (pelle agra —шершава шкіра; італ.); для пелагри властиві також патологічні зміни слизової оболонки ротової порожнини та кишечника з наявністю важкої діареї; при тривалому перебігу захворювання (починаючи з раннього дитячого віку) можливий розвиток розумового відставання (деменції). Введення препаратів нікотинової кислоти або нікотинаміду протидіє симптомам авітамінозу (звідси назва — Pellagra Preventing vitamin; англ.). Вперше хвороба була виявлена у жителів країн Середньоземноморського узбережжя в умовах харчування продуктами з кукурудзи, які містять недостатню кількість як вільного ніацину, так і триптофану, з якого може синтезуватися певна кількість нікотинаміду. Випадки пелагри спостерігалися внаслідок недостатнього харчування в’язнів в умовах концентраційних таборів. Джерела та добова потреба Нікотинова кислота та нікотинамід містяться в достатній кількості у складі поширених продуктів харчування рослинного та тваринного походження (хлібові, круп’яних виробах, овочах, м’ясних продуктах). Добова потреба у вітаміні РР складає 15-25 мг* [19 мг]**; еквівалентними 1 мг
Вітамін В6 Терміном піридоксин об’єднують три близькі за дією та взаємно перетворювані в біологічних тканинах сполуки: піридоксол (2-метил-3-окси-4,5-діоксиметил- піридин), піридоксаль та піридоксамін:Біологічні властивості та механізм дії Біологічна дія вітаміну В6 пов’язана з участю в реакціях амінокислотного обміну. Його коферментними формами є піридоксальфосфат (ПАЛФ) та піридоксамін-фосфат (ПАМФ), що беруть участь у каталітичних циклах таких ферментних реакцій: – реакцій трансамінування у складі ферментів амінотрансфераз (коферментну функцію виконують ПАЛФ та ПАМФ, що взаємно перетворюються в ході акту каталізу); – реакцій декарбоксилювання у складі ферментів декарбоксилаз амінокислот (коферментну функцію виконує ПАЛФ); – перетворення триптофану кінуреніновим шляхом з утворенням нікотинаміду НАД (в метаболічному шляху бере участь ПАЛФ-залежний фермент кінуреніназа);
Прояви недостатності піридоксину найчастіше спостерігаються в дитячому віці за умов харчування синтетичними сумішами, не збалансованими за вмістом цього вітаміну, і проявлятися пелагроподібним дерматитом, судомами, анемією. Вважають, що розвиток судом може бути пов’язаним з дефіцитом утворення в нервовій системі гальмівного медіатора ГАМК внаслідок недостатньої активностї ПАЛФ-залежної глутаматдекарбоксилази.Стан недостатності вітаміну В6 може бути також спричиненим тривалим застосуванням протитуберкульозних засобів — похідних ізонікотинової кислоти (Ізоніазид тощо), що є структурними аналогами піридоксину і можуть виступати як його антивітаміни. Джерела та добова потреба Вітамін В12 Вітамін В12 (кобаламін) за хімічною будовою належить до класу кориноїдів; його молекула складається з двох частин — кобальтвмісної порфіриноподібної (хромофорної) та нуклеотидної структур: Атом Со+, що міститься в центрі ядра хромофорної структури, в комерційних препаратх вітаміну В12 сполучений із ціанідною групою (ціанокобаламін). У разі заміщення ціаногрупи на інші радикали утворюються такі похідні вітаміну В12, як гідроксикобаламін (вітамін В12В), нітрокобаламін (вітамін В12С); в організмі людини синтезуються коферментні форми вітаміну — метилкобаламін (міститься в цитоплазмі) і 5 дезоксиаденозилкобаламін (мітохондріальна форма коферменту). Біологічні властивості та механізм дії Коферментні форми вітаміну В12 беруть участь в каталізі біохімічних реакцій такими ферментами:– метилмалоніл-КоА-мутазою (ферментом, що каталізує реакцію перетворення метилмалоніл-КоА на сукциніл-КоА); коферментом є 5-дезоксиаденозилкобаламін (механізм реакції розглянутий у главі 18). Реакція має значення для метаболізму метилмалоніл-КоА, що утворюється при розщепленні амінокислот із розгалуженими ланцюгами — L-валіну, L-лейцину, L-ізолейцину таβ-окисленні жирних кислот з непарною кількістю вуглецевих атомів; – гомоцистеїн-метилтрансферазою (в реакції синтезу метіоніну з гомоцистеїну); коферментом є метилкобаламін, що переносить метильну групу на гомоцистеїн з N5-метилтетрагідрофолату. Біохімічне значення реакції полягає в продукуванні метіоніну, який є головним донором метильних груп у реакціях синтезу фізіологічно активних сполук, метилюванні нуклеотидів нуклеїнових кислоти тощо. Недостатність у вітаміні В12 (хвороба Адисона-Бірмера; злоякісна, або пернізіозна анемія) може розвиватися за механізмом ендогенного гіпо-(а-) вітамінозу внаслідок порушення всмоктування кобаламіну в шлунково-кишковому тракті. Патологія спричиняється відсутністю синтезу в шлунку глікопротеїну транскорину (так званого “внутрішнього фактора Касла”), що є транспортером “зовнішнього фактора Касла” (тобто вітаміну В12) через слизову оболонку тонкої кишки; тканинне депо вітаміну створюється в печінці. Біохімічною основою розвитку злоякісної вітамін В12-залежної анемії є порушення біосинтезу нуклеїнових кислот і білків, що проявляється насамперед у тканинах з інтенсивною клітинною проліферацією, до яких належить кровотворна тканина. Ця форма анемії характеризується значним зменшенням кількості еритроцитів (до 1,5-2·1012/л при нормі 5·1012/л) при збільшенні їх об’єму і зміні форми клітин (макроцитарна, мегалобластична анемія); характерними симтомами хвороби є також порушення з боку периферичної нервової системи внаслідок демієлінізації нервових стовбурів (фунікулярний мієлоз), хейліт та глосит (“полірований” кінчик язика). Захворювання розвивається як наслідок атрофічного гастриту, раку шлунка, гастректомії, інвазії гельмінтом лентецем широким (Diphyllobothrium latum). Джерела та добова потреба Потреби організму людини у вітаміні В12 значною мірою забезпечуються за рахунок синтезу його мікрофлорою товстої кишки; крім того, кобаламін міститься в достатній кількості в тваринній їжі — найбільш багатим джерелом вітаміну В12 є печінка, яка містить до 100 мг вітаміну/100 г продукту. Добова потреба у вітаміні В12 складає 2-5 мкг* [2 мкг]**.
66 Фолієва кислота (ФК; фолацин; інші назви: Вітамін Вс, Вм, В9, В11) є за хімічною природою похідним птеринів — птероїлглутаміновою кислотою, що містить у складі молекули сполучені з похідним птеридину (птерином) фрагменти п-амінобензойної кислоти (ПАБК) та L-глутамату: Біологічні властивості та механізм дії Коферментною формою фолієвої кислоти є її гідроване похідне 5,6,7,8-тетрагідрофолієва кислота (ТГФК). Коферментні функції ТГФК полягають у міжмолекулярному переносі одновуглецевих фрагментів (метильного, метиленового,метенільного, оксиметильного, формільного, форміміно), що використовуються в багатьох реакція обміну амінокислот, синтезі нуклеотидів (тимідилату ДНК, пуринових ядер ДНК та РНК), фізіологічно активних сполук (глава 18). Фолієва кислота біохімічно пов’язана з обміном та функціями вітаміну В12, а саме: – ТГФК (у вигляді N5-метилтетрагідрофолату) разом із вітаміном В12 (метилкобаламіном) беруть участь у реакції синтезу метіоніну (гомоцистеїнметилтрансферазна реакція — див. вище); фізіологічне значення процесу полягає в утворенні метіоніну — донора метильних груп у метилюванні нуклеотидів нуклеїнових кислот (ДНК та певних класів РНК), синтезі холіну, креатину тощо; – хвороби недостатності обох вітамінів часто перебігають сумісно і мають близьку клінічну картину. Класичним проявом авітамінозу ФК є захворювання “спру”, що характеризується макроцитарною анемією та пінистими проносами(spruw — піна; голандськ.); захворювання розвивається внаслідок споживання раціону, збідненого білками, що призводить до порушення як синтезу мікро- організмами фолієвої кислоти, так і (в подальшому) засвоєння вітаміну В12. Джерела та добова потреба Найбільш багатими природними джерелами фолієвої кислоти є листя зелених рослин, в яких вона синтезується (звідси назва вітаміну: folium — листя; лат.). Потреби людини у вітаміні забезпечуються за рахунок синтезу його мікрофлорою кишечника, а також споживання рослинної та тваринної їжі; значна кількість ФК міститься в печінці та дріжджах. До розвитку недостатності вітаміну може призводити дисбактеріоз, спричинений тривалим прийомом сульфаніламідних препаратів, які, виступаючи структурними аналогами ПАБК (компонента молекули птероїлглутамінової кислоти), блокують утворення в бактеріальних клітинах ФК, необхідної для синтезу власних нуклеїнових кислот мікроорганізмів. Добова потреба у фолієвій кислоті становить 200-400 мкг* Вітамін Н
Біологічні властивості та механізм дії Емпірична назва вітаміну — біотин — склалася історично і свідчить про його необхідність для процесів життєдіяльності (bios — життя; грецьк.). Коферментну функцію виконує біотин, що сполучений за типом простетичної групи (через ε-аміногрупу лізинового залишку білка) з ферментами карбоксилювання. Акцептуючи діоксид вуглецю (СО2) з утворенням карбоксибіотину, вітамін Н бере участь у таких біохімічних реакціях:
|
|||||||||
|
|
Последнее изменение этой страницы: 2017-01-24; просмотров: 175; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 18.188.252.23 (0.074 с.) |
 Недостатність сахарази звичайно виявляється разом із недостатністю ізомальтази у вигляді сполученого дефекту — непереносимості двох дисахаридів. Ферментопатія проявляється після переводу новонароджених на мішане харчування з додаванням фруктових соків та інших продуктів, що містять рослинні цукри. Клінічно недостаність дисахаридаз проявляється симптомами вуглеводної диспепсії — діареєю, метеоризмом; новонароджені діти відстають у розвитку. Перетравлення ліпідів Перетравлення ліпідів харчових продуктів здійснюється в дванадцятипалій кишці під впливом ферментів, що синтезуються в неактивній формі в екзокринних клітинах підшлункової залози, а саме — ліпази, фосфоліпази А2, холестеролестерази та власних ферментів кишечника. 1. Гідроліз нейтральних жирів відбувається за рахунок дії ліпази підшлункової залози. Панкреатична ліпаза специфічна до складноефірних зв’язків в положеннях 1 та 3 триацилгліцеролів, тому продуктами дії ферменту є вільні жирні кислоти та 2-моноацилгліцерол: Активне функціонування панкреатичної ліпази реалізується за умов оптимальної лужності (рН панкреатичного секрету = 7,5-8,0) та наявності амфіпатичних молекул жовчних кислот (головним чином — глікохолевої і таурохолевої), які необхідні для емульгування харчових жирів і утворення міцел триацилгліцеролів. Процес взаємодії ферментного білка ліпази з поверхнею розподілу фаз у системі жовчно- кисла сіль/триацилгліцерол потребує також наявності додаткового фактора — білка коліпази, який міститься в секреті підшлункової залози. Крім емульгування харчових жирів, що є передумовою дії панкреатичної ліпази, жовчні кислоти беруть також участь у формуванні і всмоктуванні міцелярних структур (вільні жирні кислоти, моногліцериди), що формуються після гідролізу триацилгліцеролів. Основна маса жовчних кислот (90- 95 % їх загальної кількості) всмоктується з кров’ю v. porta в нижніх відділах тонкї кишки і надходить у печінку, де повторно використовується для формування жовчі — процесс ентерогепатичної циркуляції. Таким чином, з каловими масами щодобово виводиться лише до 0,5 г жовчних кислот; їх втрата компенсується за рахунок синтезу в гепатоцитах нових молекул первинних жовчних кислот (холевої та хенодезоксихолевої) з холестерину. 2. Гідроліз фосфоліпідів (гліцерофосфоліпідів) каталізується фосфоліпазою А2, яка синтезується в підшлунковій залозі у вигляді проферменту та перетворюється в активну форму шляхом триптичного гідролізу певних пептидних зв’язків у молекулі каталітично неактивного білка. Панкреатична фосфоліпаза А2 гідролізує складноефірні зв’язки в положенні 2 фосфогліцеридів з утворенням лізофосфоліпідів. Інші фосфоліпази, що містяться в кишковому соку, розщеплюють гліцерофосфоліпіди до гліцерину, вищих жирних кислот, азотистих основ та фосфорної кислоти: 3. Гідроліз ефірів холестерину відбувається під дією холестеролестерази (гідролази холестерилових ефірів) з утворенням холестеролу, який всмоктується ентероцитами у вільній формі. У результаті розглянутих біохімічних процесів, що відбуваються з харчовими ліпідами в порожнині кишечника, утворюється складна суміш продуктів, основними компонентами якої є: – вільні вищі жирні кислоти (у вигляді Nа+ та К+-солей); – 2-моноацилгліцероли; – вільний (неетерифікований) холестерин; – продукти гідролізу гліцерофосфоліпідів (гліцерин, аміноспирти, солі фосфорної кислоти); – триацилгліцероли, що містять залишки коротколанцюгових (переважноС8-С10) жирних кислот. Такі триацилгліцероли складають до 10 % від загальної
Недостатність сахарази звичайно виявляється разом із недостатністю ізомальтази у вигляді сполученого дефекту — непереносимості двох дисахаридів. Ферментопатія проявляється після переводу новонароджених на мішане харчування з додаванням фруктових соків та інших продуктів, що містять рослинні цукри. Клінічно недостаність дисахаридаз проявляється симптомами вуглеводної диспепсії — діареєю, метеоризмом; новонароджені діти відстають у розвитку. Перетравлення ліпідів Перетравлення ліпідів харчових продуктів здійснюється в дванадцятипалій кишці під впливом ферментів, що синтезуються в неактивній формі в екзокринних клітинах підшлункової залози, а саме — ліпази, фосфоліпази А2, холестеролестерази та власних ферментів кишечника. 1. Гідроліз нейтральних жирів відбувається за рахунок дії ліпази підшлункової залози. Панкреатична ліпаза специфічна до складноефірних зв’язків в положеннях 1 та 3 триацилгліцеролів, тому продуктами дії ферменту є вільні жирні кислоти та 2-моноацилгліцерол: Активне функціонування панкреатичної ліпази реалізується за умов оптимальної лужності (рН панкреатичного секрету = 7,5-8,0) та наявності амфіпатичних молекул жовчних кислот (головним чином — глікохолевої і таурохолевої), які необхідні для емульгування харчових жирів і утворення міцел триацилгліцеролів. Процес взаємодії ферментного білка ліпази з поверхнею розподілу фаз у системі жовчно- кисла сіль/триацилгліцерол потребує також наявності додаткового фактора — білка коліпази, який міститься в секреті підшлункової залози. Крім емульгування харчових жирів, що є передумовою дії панкреатичної ліпази, жовчні кислоти беруть також участь у формуванні і всмоктуванні міцелярних структур (вільні жирні кислоти, моногліцериди), що формуються після гідролізу триацилгліцеролів. Основна маса жовчних кислот (90- 95 % їх загальної кількості) всмоктується з кров’ю v. porta в нижніх відділах тонкї кишки і надходить у печінку, де повторно використовується для формування жовчі — процесс ентерогепатичної циркуляції. Таким чином, з каловими масами щодобово виводиться лише до 0,5 г жовчних кислот; їх втрата компенсується за рахунок синтезу в гепатоцитах нових молекул первинних жовчних кислот (холевої та хенодезоксихолевої) з холестерину. 2. Гідроліз фосфоліпідів (гліцерофосфоліпідів) каталізується фосфоліпазою А2, яка синтезується в підшлунковій залозі у вигляді проферменту та перетворюється в активну форму шляхом триптичного гідролізу певних пептидних зв’язків у молекулі каталітично неактивного білка. Панкреатична фосфоліпаза А2 гідролізує складноефірні зв’язки в положенні 2 фосфогліцеридів з утворенням лізофосфоліпідів. Інші фосфоліпази, що містяться в кишковому соку, розщеплюють гліцерофосфоліпіди до гліцерину, вищих жирних кислот, азотистих основ та фосфорної кислоти: 3. Гідроліз ефірів холестерину відбувається під дією холестеролестерази (гідролази холестерилових ефірів) з утворенням холестеролу, який всмоктується ентероцитами у вільній формі. У результаті розглянутих біохімічних процесів, що відбуваються з харчовими ліпідами в порожнині кишечника, утворюється складна суміш продуктів, основними компонентами якої є: – вільні вищі жирні кислоти (у вигляді Nа+ та К+-солей); – 2-моноацилгліцероли; – вільний (неетерифікований) холестерин; – продукти гідролізу гліцерофосфоліпідів (гліцерин, аміноспирти, солі фосфорної кислоти); – триацилгліцероли, що містять залишки коротколанцюгових (переважноС8-С10) жирних кислот. Такі триацилгліцероли складають до 10 % від загальної

 – біосинтезі гему (ПАЛФ є коферментом δ-амінолевулінатсинтази — ферменту, що каталізує утворення δ-амінолевулінату, першого метаболіту цього синтетичного шляху).
– біосинтезі гему (ПАЛФ є коферментом δ-амінолевулінатсинтази — ферменту, що каталізує утворення δ-амінолевулінату, першого метаболіту цього синтетичного шляху). Потреби у вітаміні В6 людина покриває за рахунок його споживання з такими поширеними продуктами харчування, як хліб, картопля, круп’яні вироби, м’ясо, печінка тощо; частково потреби в цьому вітаміні задовольняються за рахунок синтезу його мікрофлорою кишечника. Добова потреба в піридоксині складає 2-3 мг*
Потреби у вітаміні В6 людина покриває за рахунок його споживання з такими поширеними продуктами харчування, як хліб, картопля, круп’яні вироби, м’ясо, печінка тощо; частково потреби в цьому вітаміні задовольняються за рахунок синтезу його мікрофлорою кишечника. Добова потреба в піридоксині складає 2-3 мг*
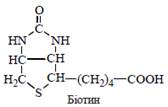 За будовою молекули вітамін Н (біотин) є продуктом конденсації сечовини та тіофенвалер’янової кислоти; його хімічна назва: 2-кето-3,4-імідазолідо-2-тетрагідротіофенвалер’янова кислота:
За будовою молекули вітамін Н (біотин) є продуктом конденсації сечовини та тіофенвалер’янової кислоти; його хімічна назва: 2-кето-3,4-імідазолідо-2-тетрагідротіофенвалер’янова кислота:


