
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Катализ в химических реакциях
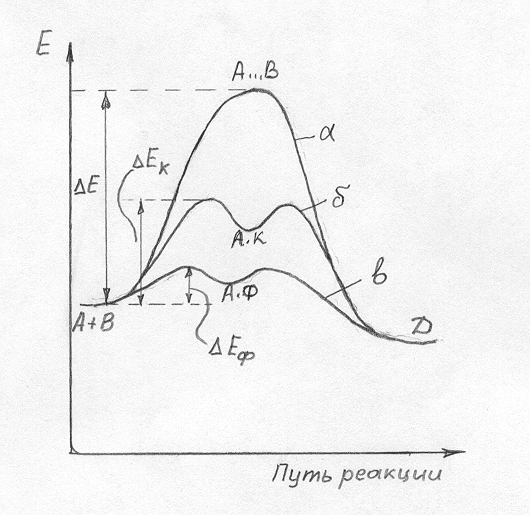
Одним из наиболее распространенных методов регулирования скоростей реакций является применение катализаторов. Катализаторы – это вещества, которые активно участвуют в промежуточных стадиях реакции, изменяют скорость суммарного процесса, но в продуктах реакции обнаруживаются в неизменном состоянии. Изменение скорости реакции в присутствии катализаторов называется катализом, а сами реакции – каталитическими реакциями. Существует два подхода к классификации каталитических реакций. 1. По наличию границы раздела фаз различают: – гомогенный катализ, когда реагенты, катализатор и продукты реакции находятся в объеме одной фазы; – гетерогенный катализ, когда катализатор и реагирующие вещества с продуктами реакции находятся в различных фазах; часто катализатор образует твердую фазу, а реагенты и продукты находятся в жид-кой фазе или в газовой фазе. 2. По характеру изменения скорости реакции бывает: – положительный катализ, при котором катализатор увеличивает скорость реакции; – отрицательный катализ (ингибирование), при котором катализатор (ингибитор) замедляет скорость реакции; – автокатализ, когда роль катализатора играет продукт реакции; например, при гидролизе сложного эфира СН3СООСН3 + Н2О образующаяся в результате реакции уксусная кислоты отщепляет ион водорода, который начинает играть роль катализатора реакции гидролиза. Поэтому сначала медленно протекающая реакция со временем имеет все более увеличивающуюся скорость. Для объяснения механизма каталитических реакций предложена теория промежуточных соединений. Согласно этой теории при положительном катализе катализатор (К) с большой скоростью образует с одним из реагентов промежуточное соединение, которое также быстро взаимодействует со вторым реагеном: А + В 1) А + К 2) АК + В Из рисунка 4а видно, что энергия активации некаталитического процесса намного больше, чем энергии активации первой и второй стадий каталитического превращения. Таким образом, при положительном катализе роль катализатора заключается в снижении энергии активации реакции.
Рисунок 4 Энергетические диаграммы каталитической реакции (а) и
ингибированной реакции (б)
В реакциях ингибирования ингибитор (I) с высокой скоростью образует прочное промежуточное соединение (АI), которое очень медленно превращается в продукт реакции: А + В 1) А + I 2) AI + В Из рисунка 4б видно, что первая стадия ингибирования по сравнению с неингибированным процессом имеет более низкую энергию активации и протекает очень быстро. В то же время энергия активации второй стадии ингибирования намного больше, чем неингибированной реакции. Таким образом, в ингибированных реакциях роль ингибитора заключается в увеличении энергии активации реакции. ОСОБЕННОСТИ ФЕРМЕНТАТИВНОГО КАТАЛИЗА
Ферменты (от лат. fermentum – закваска) – биологические катализаторы, присутствующие во всех биологических системах. Они осуществляют превращения веществ в организме, тем самым направляя и регулируя в нем обмен веществ. Ферменты находят широкое применение в пищевой и легкой промышленности. По химической природе ферменты представляют собой молекулу глобулярного белка. Ферментативный катализ (биокатализ) – это ускорение химических реакций в биологических системах специальными белками – ферментами. В основе ферментативного катализа лежат те же химические закономерности, что и в основе обычного химического катализа, используемого в химическом производстве. Вместе с тем ферментативный катализ имеет свои особенности: 1. Более высокая активность по сравнению с химическими катализаторами (увеличение скорости в 1010 – 1013 раз). Это происходит потому, что ферментативные реакции на всех стадиях имеют очень низкие энергии активации (рисунок 5).
2. Большинство ферментов отличаются специфичностью действия, так что практически каждая реакция превращения реагирующего вещества (субстрата) в продукт осуществляется специальным ферментом. Существуют две теории специфичности действия ферментов: 1) теория Фишера (теория «ключа–замка»): фермент и субстрат по пространственному строению должны подходить друг к другу как ключ к своему замку;
2) теория Кошланда (теория «руки и перчатки»): фермент и субстрат в отдельности могут не иметь соответствующие друг к другу пространственные формы, но при сближении конфигурации их изменяются таким образом, чтобы стало возможным строгое пространственное соответствие. 3. Ферментам свойственно явление инактивации – разрушение молекулы фермента после взаимодействия с определенным числом молекул субстрата. Чем выше активность фермента, тем быстрее он разрушается. Явление инактивации объясняет теория Кошланда. Действительно, чем активнее фермент, тем интенсивнее он взаимодействует с субстратом, при котором молекула фермента претерпевает значительную пространственную деформацию. Такая многократная деформация приводит к разрыву наиболее слабых химических связей, то есть к разрушению молекулы фермента. 4. Каждый фермент содержит молекулу белка. Однокомпонентные состоят только из молекулы белка, а двухкомпонентные – из молекулы белка и связанного с ней небелкового компонента (неорганического иона или молекулы органического соединения – чаще всего молекулы витамина или продукта его превращения) – кофактора. Молекулярный комплекс белка и кофактора называется холоферментом, который обладает максимальной каталитической активностью. В составе холофермента белковая часть называется ферон, а небелковая часть – агон. Белковый компонент, лишенный кофактора называется апоферментом, а кофактор, отделенный от белковой молекулы – коферментом. Отдельно от кофактора молекула белка обладает очень низкой активностью, а кофермент как катализатор вообще неактивен. 5. Действие большинства ферментов регулируется, то есть они способны переходить от состояния с низкой активностью к состоянию с высокой активностью и обратно. Механизм регуляции представляет сложную систему, с помощью которого организм контролирует все свои функции. 6. Ферменты очень чувствительны к влиянию внешних условий. Они проявляют активность в относительно узком диапазоне температур и значения рН среды. Механизм ферментативных реакций аналогичен механизму реакций, катализируемых химическими катализаторами: S + E то есть сначала очень быстро образуется ферментсубстратный комплекс ES, который может обратно диссоциировать на субстрат S и фермент Е, но и равным образом медленно превращаться в продукт реакции P. При постоянстве концентрации фермента зависимость начальной скорости превращения субстрата v0 от его начальной концентрации v0 = где Km и Vmax – кинетические параметры, отражающие механизм действия фермента. Методика определения этих параметров основана на использовании уравнения Лайнуивера – Берка, которое получается путем преобразования уравнения Михаэлиса – Ментен:
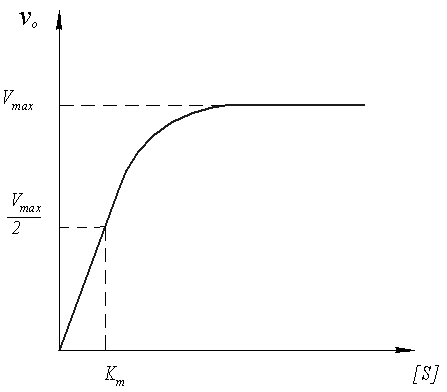
На рисунке 6 показана методика определения параметров Km и Vmax. Vmax - это максимальная начальная скорость реакции при данной концентрации фермента [ E ] (рисунок 7). Молярная активность фермента (аЕ) определяется соотношением:
аЕ = которое показывает количество молекул субстрата, превращаемого одной молекулой фермента за единицу времени. Например, для реак-ции СО2 + Н2О
Рисунок 7 Зависимость начальной скорости ферментативной реакции от начальной концентрации субстрата
Параметр Km имеет смысл количества субстрата, необходимого для связывания половины имеющегося фермента в фермент-субстратный комплекс и достижения половины максимальной скорости (рисунок 7). Поэтому Km можно использовать для оценки специфичности действия определенного фермента по отношению к данному субстрату. Например, для реакции моносахарид + АТФ катализируемой ферментом гексокиназой, для глюкозы получено Кm = 8∙10–6 моль/л, а для аллозы Кm = 8∙10–3 моль/л. Следовательно, фермент более предпочтительно взаимодействует с глюкозой, так как для достижения одного и того же результата его требуется в 1000 раз меньше, чем аллозы.
4. ХИМИЧЕСКОЕ РАВНОВЕСИЕ При достижении химически равновесного состояния число молекул веществ перестает меняться и остается постоянным во времени при неизменных внешних условий. Для химического равновесия характерны следующие признаки: 1) равенство скоростей прямой и обратной реакций; 2) постоянство концентраций (парциальных давлений) компонентов при постоянстве внешних условий; 3) подвижность, то есть способность самопроизвольно восстанавливаться при небольших смещениях; 4) равновесие достигается как прямым, так и обратным течением реакции. Рассмотрим энергетическую диаграмму химической реакции А + В
Рисунок 8 Энергетическая диаграмма обратимой химической реакции Следовательно, при данной температуре прямая и обратная реакции имеют вполне определенные значения константы скорости. Поэтому в обратимых реакциях кинетические кривые имеют вид, приведенный на рисунке 9 а. Из рисунка видно, что после достижения времени tр концентрации компонентов остаются неизменными. Согласно закон у действия масс
Из рисунка 9 б видно, что после достижения времени установления равновесия tp достигается равенство скоростей
Kc где Kc =
Рисунок 9 Кинетические кривые (а) и зависимости скорости прямой и обратной реакций от времени (б) для обратимой реакции В общем случае для реакции mA +nB константа равновесия определяется выражением Kc Таким образом, Kc - это параметр, характерный для реакционной системы при данной температуре, определяющий соотношение концентраций компонентов в состоянии химического равновесия. Если реакция протекает в газовой фазе, то вместо концентраций используют парциальные давления компонентов системы. Для приведенной выше равновесной реакции константу равновесия, определенную по парциальным давлениям компонентов в состоянии равновесия, находят как Kр Для идеальных газов рi=Ci RT. Поэтому
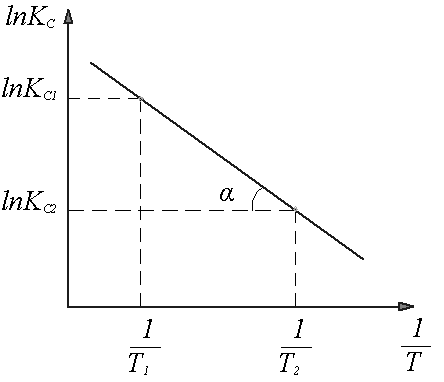
где Значения Kc и Kp зависят от температуры и от природы компонентов реакционной систем. Из уравнений Аррениуса для прямой и обратной реакции следует: ln kпр = ln Aпр Откуда ln Так как ln Kр = ln где ΔНпр – тепловой эффект прямой реакции. Из полученного уравнения следует, что зависимость Kp Для определения ΔHпр аналитическим методом находят значение Kp при двух разных температурах и проводят вычисления по формуле ΔHпр
Рисунок 10 Определение теплового эффекта прямой эндотермической реакции (ΔНпр >0) Последнее выражение называется интегральным уравнением изобары химической реакции. Она связывает константы равновесия при двух различных температурах и описывает равновесные системы, в которых при изменении температуры общее давление остается постоянным. Если при изменении температуры объем системы сохраняется постоянным, как, например, при реакциях в растворах, то взаимосвязь параметров выражается через изохору химической реакции ΔUпр Обсуждая направление протекания химических реакций с точки зрения химической термодинамики было отмечено, что система находится в состоянии химического равновесия при условии ∆G= 0. Исходя из этого положения получено уравнение изотермы химической реакции, которая позволяет определять знак ∆G и, соответственно, направление химической реакции при условии смешения компонентов реакционной системы в произвольных соотношениях: ΔG = RT (ln где pA и рВ - произвольные парциальные давления компонентов, получаемые при их смешивании. Аналогичное соотношение предложено и для системы, компоненты которой находятся в растворе. Например, для реакции mA+nB равновесие которой устанавливается в жидкой фазе, уравнение изотермы химической реакции имеет следующий вид: ΔG = RT (ln где Смещение равновесия. Изменение температуры, концентрации, давления системы, находящейся в состоянии равновесия, выводит ее из равновесия. Но через определенное время в системе снова устанавливается новое равновесное состояние, параметры которой уже отличаются от первоначального состояния. Такой переход системы из одного равновесного состояния в другое равновесное состояние при изменении условий называется смещением равновесия. Его используют с целью увеличения выхода целевого продукта для тех систем, которые имеют небольшие значения констант равновесия. Кроме того, методом смещения равновесия можно подавлять параллельно протекающие нежелательные процессы.
Но при этом необходимо иметь в виду два фактора, которые не влияют на состояние равновесия. Во-первых, ввод катализатора в равноваесную систему не приводит к смещению равновесия. Катализатор одновременно понижает энергию активации прямой и обратной реакции, что приводит к повышению скорости обеих реакций в одинаковой степени. В результате применения катализатора состояние равновесия достигается за более короткий промежуток времени. Во-вторых, в гетерогенных равновесных системах концентрации и парциальные давления нерастворимых и нелетучих твердых веществ не входят в выражение константы равновесия. Например, для реакции FeO +CO Влияние температуры. Уравнения изохоры и изобары позволяют предсказывать направление смещения равновесия при изменении температуры. Например, если система в равновесии и прямая реакция экзотермическая (DНпр <0), то при повышении температуры (Т2>Т1) должно соблюдаться неравенство Kp,2 Повышение температуры смещает равновесие в сторону протекания эндотермической реакции, а понижение температуры – в сторону экзотермической реакции. Таким образом, наибольший выход продуктов достигается: - для экзотермических реакций при низких температурах; - для эндотермических реакций при высоких температурах. Влияние концентрации (парциального давления). Уравнение изотермы позволяет предсказывать направление смещения равновесия при изменении концентрации какого-либо компонента равновесной системы. Пусть система находится в состоянии равновесия. Тогда ΔG =0 и концентрации компонентов в уравнении изотермы соответствуют равновесным значениям и Увеличение концентрации (парциального давления) исходных реагентов смещает равновесие в сторону образования продуктов, а уменьшение их концентрации (парциального давления) – в сторону обратного превращения продуктов в исходные. Увеличение концентрации(парциального давления) продуктов смещает равновесие в сторону обратной реакции, а уменьшение их концентрации(парциального давления) – в сторону прямой реакции. Поэтому для увеличения выхода продукта реакции необходимо увеличить концентрации (парциальные давления) исходных реагентов или же уменьшить концентрацию (парциальные давления) продуктов путем постепенного вывода их из реакционной системы. Влияние общего давления системы. Пусть дана равновесная газофазная система mA Согласно закону Дальтона, pA = p∙yA и pB = p∙yB, где р - общее давление в системе; рА, рВ – парциальные давления компонентов; yA, yB – мольные доли компонентов в газовой фазе. Тогда уравнение изотермы принимает следующий вид
или
Если при давлении р1 система находится в равновесии, то
Повышение давления до р2 выводит систему из равновесия. Так как (п-т)
Это термодинамическое условие протекания обратной реакции. Следовательно, при повышении давления новое равновесное состояние возникнет в результате обратного превращения продукта В в исходное соединение А, в результате чего уменьшается общее число молекул в системе. Обобщая полученный результат можно сделать следующие выводы: - повышение общего давления системы смещает равновесие в сторону той реакции, которая идет с уменьшением числа молекул; - понижение общего давления системы приводит к смещению равновесия в сторону той реакции, которая протекает с увеличением числа молекул. Обобщение закономерностей влияния всех факторов на направление смещения равновесия приводит к правилу, которое называется принципом Ле-Шателье: если на равновесную систему оказать внешнее воздействие (изменить температуру, концентрацию или парциальные давления компонентов, общее давление), то она отреагирует таким образом, чтобы эффект этого воздействия был ослаблен. ФОТОХИМИЧЕСКИЕ РЕАКЦИИ
Химические реакции, протекающие под воздействием светового излучения, называются фотохимическими реакциями. К наиболее важным фотохимическим реакциям относятся реакции образования озона из молекулярного кислорода под действием ультрафиолетовой радиации Солнца: О2 + h O Образующийся при этом озон О3 поглощает ультрафиолетовые лучи в области 250-260 ммк, которые губительно действуют на живые организмы. К другой важной фотохимической реакции относится фотосинтез, в результате которого происходит поглощение растениями углекислого газа из атмосферы и выделение кислорода. Фотохимическое разложение бромида серебра лежит в основе фотографического процесса. Энергия фотона (кванта излучения) (Е) определяется соотношением E = h где h – постоянная Планка (h 1. Атом или молекула переходят в возбужденное состояние: А + h М + h 2. Диссоциация молекулы с образованием атомов или свободных радикалов: АВ + h 3. Образование простых или молекулярных ионов путем отрыва одного электрона: А + h AB + h Все эти процессы подчиняются следующим законам. 1. Фотохимические реакции могут быть вызваны только той частью падающего излучения, которая поглощается реагирующей системой (закон Гротгуса - Дрепера). 2. Каждый поглощенный квант излучения вызывает превращение только одной молекулы (закон Эйнштейна - Штарка). 3. Количество образующегося в результате фотохимической реакции продукта пропорционально интенсивности поглощенного излучения и времени облучения (закон Вант-Гоффа). Последний закон можно представить в математической форме: m = k где m – масса фотохимически превращенного вещества, г; При опытной проверке 1-го и 2-го законов иногда наблюдается кажущееся несоответствие. Во-первых, число поглощенных квантов бывает не равно числу прореагировавших молекул вещества, т.е. как бы нарушается закон Эйнштейна-Штарка. Поэтому для характеристики фотохимических процессов введено понятие квантового выхода Во-вторых, некоторые вещества не поглощают света в видимой или ультрафиолетовой области, тем не менее, они способны претерпевать превращение при облучении. Таким образом, как бы нарушается закон Гротгуса. Оказалось, что в этом случае квант излучения поглощается особыми веществами – фотосенсибилизаторами, которые передают поглощенную энергию другому веществу, которое и претерпевает в результате этого химическое превращение. Следовательно, нарушение закона Гротгуса является лишь кажущимся. Например, молекулярный водород не поглощает световое излучение с длиной волны 253,7 нм. Однако при облучении смесь паров ртути и водорода, наблюдается процесс диссоциации молекул водорода на атомы: Hg + h Hg* + H2 Аналогичным фотосенсибилизированным процессом является фотосинтез – синтез углеводов из оксида углерода (IV) и воды, сопровождающийся выделением кислорода. В качестве сенсибилизатора данной фотохимической реакции выступает молекула хлорофилла. Причем хлорофилл b улавливает и собирает энергию светового излучения. После фотовозбуждения он передает избыточную энергию молекуле хлорофилла а, которая затем принимает непосредственное участие в процессе фотосинтеза. Суммарный процесс фотосинтеза выражается реакцией: 6СО2 + 6Н2О Фотосинтез – сложный окислительно-восстановительный про-цесс, сочетающий фотохимические реакции с ферментативными. В механизме фотосинтеза различают две стадии – световую и темно-вую. Световая стадия включает собственно фотохимические реакции и сопряженные ими ферментативные, которые завершают окисление воды и образуют восстановленный никотинамид-адениндинуклеотид-фосфат (НАДФН2) и аденозинтрифосфорную кислоту (АТФ). В темновой стадии НАДФН2 и АТФ восстанавливают молекулу СО2 до СН2О и далее образуется моносахарид в цикле сопряженных ферментативных реакций, которые идут без участия кванта излучения.
СВОЙСТВА РАСТВОРОВ ОБЩИЕ СВЕДЕНИЯ
Растворами называются гомогенные (однофазные) системы, состоящие из растворителей, раствореннных веществ и продуктов их взаимодействия, концентрации которых могут изменяться в широких пределах. Они могут быть твердыми, жидкими и газообразными. Процессы в биологических объектах и технологические процессы в перерабатывающей промышленности сельского хозяйства протекают в водных растворах. Поэтому в дальнейшем ограничимся рассмотрением только водных растворов различных веществ. При растворении происходит равномерное распределение молекул или ионов растворяемого вещества в объеме растворителя. Однако растворение нельзя рассматривать как чисто физический процесс диффузии одного вещества в другом. Об этом свидетельствует выделение значительного количества теплоты при растворении в воде некоторых веществ (H2SO4, NaOH и другие). Установлено, что между молекулами растворителя и молекулами или ионами растворенного вещества возможны химические взаимодействия, сопровождающиеся разрывом одних и образованием других химических связей. Это приводит к образованию продуктов взаимодействия растворителя с растворенным веществом, которые называются сольватами, а в водных растворах – гидратами. Сам процесс взаимодействия соответственно называется сольватацией или гидратацией. В настоящее время растворы рассматриваются как физико - химические системы, занимающие по своим свойствам промежуточное положение между механическими смесями и химическими соединениями, и имеют характерные для них физико-химические закономерности. Основной характеристикой всякого раствора является его концентрация. Как правило, растворителем выступает тот компонент раствора, который содержится в относительно большем количестве и определяет его фазовое состояние. Физико-химические свойства растворов зависят от их концентраций. Имеется множество таких зависимостей. Все они получены исходя из предположения, что раствор является идеальным. Идеальным называется такой раствор, в котором: 1) очень низка концентрация растворенного вещества – мольная доля менее 0,005; 2) растворенное вещество является нелетучим, то есть его молекулы не могут выйти из жидкой фазы в газовую фазу; 3) отсутствуют силы взаимодействия между частицами раствора, то есть теплота смешения равна нулю (
|
||||||||||||||||||
|
|
Последнее изменение этой страницы: 2017-01-26; просмотров: 624; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.138.141.202 (0.539 с.) |
 СН3СООН + СН3ОН
СН3СООН + СН3ОН


 ES
ES  описывается кинетическим уравнением Михаэлиса - Ментен:
описывается кинетическим уравнением Михаэлиса - Ментен: ,
, =
=  +
+ 

 ,
,
 D (рисунок 8). Для этой реакции:
D (рисунок 8). Для этой реакции:




 . Тогда
. Тогда

 - константа химического равновесия, определенная по равновесным концентрациям компонентов.
- константа химического равновесия, определенная по равновесным концентрациям компонентов.
 qD +fE
qD +fE


 -
-  - изменение количество молей компонентов в ходе реакции.
- изменение количество молей компонентов в ходе реакции. и ln kобр = ln Aобр
и ln kобр = ln Aобр 
 ln
ln 
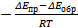
 , то
, то
 имеет вид прямой линии и для нее
имеет вид прямой линии и для нее  (рисунок 10), откуда следует
(рисунок 10), откуда следует  .
. 
 .
.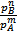 – ln Kp)
– ln Kp) qD+fE,
qD+fE, - ln Kc)
- ln Kc) - мольные доли компонентов в растворе, который получен путем смешения произвольного количества веществ А, В, D и Е.
- мольные доли компонентов в растворе, который получен путем смешения произвольного количества веществ А, В, D и Е. Fe +CO2 константу равновесия определяют как Kp =
Fe +CO2 константу равновесия определяют как Kp =  .
. Kp,1. Это говорит о том, что в новом равновесном состоянии парциальное давление продуктов реакции будет меньше, то есть реакция смещается влево.
Kp,1. Это говорит о том, что в новом равновесном состоянии парциальное давление продуктов реакции будет меньше, то есть реакция смещается влево. nB, для которой n
nB, для которой n  m, то есть прямая реакция идет с увеличением числа молекул.
m, то есть прямая реакция идет с увеличением числа молекул.
 (4.16)
(4.16) .
.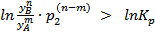 и ΔG> 0.
и ΔG> 0.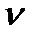

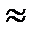 6,626
6,626  10
10  Дж∙с);
Дж∙с);  . Длина волны видимого света, инфракрасных и ультрафио-летовых лучей лежит в интервале от 100 нм до 1000 нм, а энергия их – от 120 кДж/моль до 1200 кДж/моль. Квант излучения поглощается одним единственным электроном атома в молекуле, вследствие чего этот электрон переходит на более высокий энергетический уровень. В результате возможны три различных последствия поглощения энергии в виде излучения:
. Длина волны видимого света, инфракрасных и ультрафио-летовых лучей лежит в интервале от 100 нм до 1000 нм, а энергия их – от 120 кДж/моль до 1200 кДж/моль. Квант излучения поглощается одним единственным электроном атома в молекуле, вследствие чего этот электрон переходит на более высокий энергетический уровень. В результате возможны три различных последствия поглощения энергии в виде излучения: + B
+ B 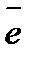

 t,
t, , который равен отношению числа действительно прореагировавших молекул к числу поглощенных квантов. Величина
, который равен отношению числа действительно прореагировавших молекул к числу поглощенных квантов. Величина  + H
+ H  С6Н12О6 + 6Н2О,
С6Н12О6 + 6Н2О,  G 0 = 2861,9 кДж/моль
G 0 = 2861,9 кДж/моль


