
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Лабораторная работа № 3. Кислотно-основное титрованиеСтр 1 из 4Следующая ⇒
ГРАВИМЕТРИЯ
Лабораторная работа № 1
Цель работы. Определение воды в твердых веществах – один из наиболее важных случаев определения, так как вода нередко является основной частью анализируемых веществ. Содержание воды определяют как прямыми, так и косвенными методами. Кристаллизационной называется вода, входящая в структуру кристаллов некоторых веществ, называемых кристаллогидратами. Содержание кристаллизационной воды в кристаллогидратах отвечает определенным химическим формулам, например ВаС12×2Н2О, CuSO4×5H2O, Na2SO4×10H2O, Na2SO4×7H2O и т. д. Вследствие этого кристаллизационную воду называют также стехиометрической водой. При нагревании кристаллогидраты разлагаются с выделением воды. На этом основано определение содержания кристаллизационной воды в большинстве кристаллогидратов методом отгонки. Так, в рассматриваемом случае навеску ВаС12×2Н2О, помещенную в бюкс, нагревают до 120 – 125 °С в сушильном шкафу до тех пор, пока не перестанет изменяться масса вещества (высушивание до постоянной массы). Достижение постоянной массы свидетельствует, очевидно, о том, что вся кристаллизационная вода уже удалена. Масса ее равна убыли в массе вещества.
Приборы и реактивы. Сушильный шкаф; технические весы; аналитические весы; эксикатор; тигельные щипцы; стеклянный бюкс; хлорид бария кристаллический, BaCl2×2H2O.
Выполнение работы. Взятие навески. 1. Тщательно вымытый бюкс высушивают в сушильном шкафу и, не закрывая крышкой, охлаждают, поставив на 20 мин в эксикатор около весов. 2. После этого, соблюдая все правила, точно взвешивают бюкс вместе с крышкой на аналитических весах. Затем помещают в него около 1,5 г свежеперекристаллизованного ВаС12×2Н2О и, закрыв крышкой, снова точно взвешивают. Высушивание. 1. Открыв бюкс, помещают крышку сверху бюкса боком (т. е. перевернув на ребро) и ставят на полку (не на дно) сушильного шкафа. Под бюкс следует подложить листок бумаги с указанием своей фамилии. Выдерживают бюкс в шкафу при температуре около 125 °С приблизительно 2 ч. Затем тигельными щипцами переносят бюкс и крышку его в эксикатор. 2. Продержав эксикатор 20 мин около весов, вынимают из него бюкс и, закрыв крышкой, точно взвешивают. Далее снова ставят бюкс с веществом в сушильный шкаф и выдерживают его там (открыв крышку) еще около 1 ч, повторно охлаждают в эксикаторе и снова взвешивают.
3. Если второе взвешивание дает тот же самый результат, что и первое, или отличается от него не более чем на 0,0002 г, кристаллизационную воду можно считать удаленной практически полностью. В противном случае высушивание с периодическим взвешиванием повторяют до тех пор, пока не будет достигнуто постоянство массы. Результаты всех повторных взвешиваний обязательно записывают в лабораторный журнал, даже если они одинаковые. Если работу приходится прерывать, бюкс оставляют в эксикаторе.
Таблица. Результаты определения кристаллизационной воды
Расчеты. Масса кристаллизационной воды в навеске: m(H2O) = 9,5895 - 9,3748 = 0,2147 г Массовая доля кристаллизационной воды в ВаС12×2Н2О вычисляют из пропорции: в 1,4575 г ВаС12×2Н2О содержится 0,2147 г Н2О в 100 г ВаС12×2Н2О – х г Н2О
Расчет ошибки определения. Для проверки найденную величину сравнивают с теоретически вычисленным значением массовой доли Н2О в ВаС12×2Н2О. В 1 моль (244,3 г) ВаС12×2Н2О содержится 2 моль воды, т.е. 36,03 г Н2О. Составим пропорцию: в 244,3 г ВаС12×2Н2О содержится 36,03 г Н2О в 100 г ВаС12×2Н2О – х г Н2О
Абсолютная ошибка составляет: d = 14,73% – 14,75% = –0,02%. Относительную ошибку определения вычисляют по формуле
Эта погрешность объясняется ошибками взвешивания. При правильной работе абсолютная ошибка данного определения не должна превышать ±0,05%. Другими словами, приемлемы результаты, лежащие в пределах от 14,70 до 14,80%.
Лабораторная работа № 2
Метод основан на осаждении сульфат-ионов в виде кристаллического белого осадка BaSO4 и получении гравиметрической формы (BaSO4) прокаливанием: SO42– + Ba2+ = BaSO4
Данным методом определяют сульфатную серу, а также свободную, пиритную и сульфидную, предварительно окислив их до сульфатной. Осаждению сульфат-ионов в виде BaSO4 мешают анионы SiO32–, SnO32–, WO42– и другие, образующие осадки соответствующих кислот при подкислении раствора. Осаждению мешают также ионы Fe3+, Al3+, С1–, МпО4– и другие, соосаждаемые с BaSO4. Мешающие ионы должны быть предварительно удалены из анализируемого раствора. Приборы и реактивы. Муфельная печь; аналитические весы; водяная или песчаная баня; эксикатор; стеклянные палочки; стеклянные воронки; фарфоровые тигли; мерный цилиндр вместимостью 100 мл; щипцы тигельные; химические стаканы вместимостью 200 – 250 и 100 мл; фильтры беззольные «синяя лента»; хлорид бария, 3 М раствор; хлороводородная кислота, 6 М раствор.
Выполнение работы. Подготовка тиглей. 1. Фарфоровые тигли моют, ополаскивают дистиллированной водой, сушат, помещают в муфельную печь и прокаливают при температуре 800 оС 15 – 20 мин. 2. Тигельными щипцами достают тигли и выставляют их для охлаждения на керамическую подставку на 1 – 2 мин. Затем тигли помещают в эксикатор до полного охлаждения. После этой операции тигли разрешается брать и переносить только тигельными щипцами или пинцетом. 3. Прокаленные и охлажденные тигли взвешивают на аналитических весах. Результаты записывают в таблицу:
Таблица. Доведение тиглей до постоянной массы
4. После взвешивания тигли снова прокаливают, охлаждают и взвешивают. Если вторая масса отличается от первой не более чем на 0,0002 – 0,0004 г, то тигли доведены до постоянной массы. В противном случае прокаливание повторяют еще раз и т. д. Получение осаждаемой формы. 1. Аликвотную часть анализируемого раствора переносят в химический стакан вместимостью 200 – 250 мл и приливают 100 мл дистиллированной воды. К полученному раствору добавляют 5 мл 2 М раствора HCl и нагревают почти до кипения на песчаной бане. 2. В химический стакан вместимостью 100 мл помещают 50 мл дистиллированной воды и 6 мл 3 М раствора ВаС12, нагревают почти до кипения. 3. Осторожно, по каплям, при постоянном перемешивании стеклянной палочкой приливают нагретый раствор ВаС12 к горячему раствору сульфата, при этом палочка не должна касаться стенок стакана. Стакан с образовавшимся осадком ставят на баню. 4. Как только осадок осядет, и жидкость над ним просветлеет, проводят проверку полноты осаждения BaSO4. Для этого к раствору над осадком приливают 2 – 3 капли горячего раствора ВаCl2, осторожно, не взмучивая осадка. Если при этом не наблюдается помутнения раствора, полнота осаждения достигнута, в противном случае приливают еще 1 мл горячего раствора ВаС12, дают осадку осесть и снова проверяют полноту осаждения. Затем стакан ставят, нагревают 2 – 3 ч для созревания осадка (или оставляют в рабочем шкафу до следующего занятия), после чего осадок фильтруют и промывают. Отделение осаждаемой формы. 1. Для фильтрования применяют бумажный беззольный фильтр «синяя лента». Подготовку фильтра см. Примечание 1. 2. Готовят промывную жидкость, в стакан на 150 мл приливают 100 мл дистиллированной воды и подкисляют 2 М раствором НС1 (8 – 10 капель на 100 мл воды).
3. Воронку с фильтром вставляют в кольцо штатива над стаканом так, чтобы оттянутый конец воронки касался внутренней стенки стакана, что ускоряет фильтрование (рис. 1). 4. Раствор, находящийся над осадком, переносят на фильтр способом декантации (см. Примечание 2). 5. После сливания большей части раствора осадок промывают, также используя способ декантации (см. Примечание 3). Для этого к осадку в стакане приливают небольшими порциями промывную жидкость. Промывную жидкость перемешивают с осадком в стакане, давая каждый раз отстояться осадку, и сливают жидкость с осадка на фильтр, как указано выше. Эту операцию повторяют 2 – 3 раза. Промывание декантацией значительно эффективнее, чем промывание на фильтре. 6. Затем осадок количественно переносят на фильтр, для чего к нему приливают небольшими порциями промывную жидкость, взмучивают осадок стеклянной палочкой и сливают суспензию на фильтр. 7. Оставшийся в стакане осадок смывают из промывалки струей промывной жидкости на фильтр, тщательно удаляют частицы осадка со дна и стенок стакана, стирая их стеклянной палочкой с резиновым наконечником. Стакан и палочку ополаскивают промывной жидкостью, сливая ее на фильтр. Частицы осадка со дна и стенок стакана снимают небольшим кусочком влажного беззольного фильтра с помощью стеклянной палочки, присоединяя этот кусочек к осадку на фильтре.
Рис. 1. Отделение осаждаемой формы: а) промывка декантацией; б) перенос осадка.
8. Для проверки полноты промывания осадка несколько капель фильтрата отбирают на часовое стекло и проводят качественную реакцию на ионы хлорида, от которых осадок отмывается. Промывание заканчивают при отрицательном результате пробы. Получение весовой формы. 1. Влажный фильтр с осадком подсушивают перед помещением в тигель. Для этого воронку с фильтром накрывают листом бумаги, проколотым в нескольких местах, и помещают в сушильный шкаф при температуре 100 °С. Необходимо следить за тем, чтобы не пересушить фильтр (ломкость бумаги). 2. Подсушенный фильтр с осадком осторожно вынимают из воронки, складывают, и переносят в тигель, масса которого доведена до постоянной. 3. Тигель устанавливают слегка наклонно в фарфоровый треугольник и небольшим пламенем газовой горелки озоляют фильтр так, чтобы он не загорелся. 4. По окончании озоления осадок в тигле прокаливают в муфельной печи при температуре 800 оС до получения постоянной массы тигля с гравиметрической формой (см. Приложение 4). Результаты прокаливания записывают в лабораторный журнал.
Таблица. Масса тиглей с осадком на аналитических весах после прокаливания
Расчеты
1. По результатам анализа рассчитать массу сульфата (SO4) в пробе. Составим пропорцию:
в 1 моль BaSO4 (233,4 г) содержится 1 моль SO4 (96,06 г) в m г «х г
2. Рассчитать относительную ошибку определения. Для этого получить у преподавателя истинное значение содержания сульфата – mист:
Примечания 1. Основная цель этой отделения осаждаемой формы – количественное отделение осадка от раствора и его очистка от адсорбированных загрязнений. Осадок отделяют от раствора путем фильтрования через бумажный беззольный фильтр или фильтрующий тигель. Бумажные беззольные фильтры изготовляют из фильтровальной бумаги, обработанной, например, НС1, HF, и применяют в том случае, если продукты горения бумаги и уголь не будут оказывать влияния на состав осадка. Масса золы беззольных фильтров менее 0,1 мг, т. е. при взвешивании на обычных аналитических весах такая масса золы не скажется на результатах взвешивания. Беззольные фильтры выпускают с различным размером пор. По плотности (пористости) фильтры маркируют цветной бумажной лентой, которой оклеивают упаковку фильтров: синяя лента – для мелкозернистых осадков типа BaSO4; белая лента – для среднезернистых типа CaCrO4 красная лента – для крупнозернистых и аморфных осадков типа Fe(OH)3. На этикетках указана зольность фильтров, обычно 0,00005 г. Размер фильтра выбирают в зависимости от количества полученного осадка, осадком должна быть заполнена одна треть фильтра. Размер воронки подбирают так, чтобы расстояние между краем фильтра и краем воронки было не меньше 5 мм и не более 15 мм. Фильтр складывают пополам, приглаживают и затем складывают еще раз примерно пополам. Затем разворачивают фильтр так, чтобы получился конус (отгибая одну створку образовавшегося уголка), и вкладывают его в стеклянную воронку так, чтобы он соответствовал по форме конусу воронки. Смачивают фильтр водой из промывалки и расправляют его, чтобы он плотно прилегал к воронке, не имел складок. 2. Для промывки декантацией на фильтр осторожно по стеклянной палочке, приставленной к носику стакана, сливают раствор, находящийся над осадком, стараясь не взмучивать осадок. Стеклянную палочку держат вертикально в левой руке, нижний конец ее должен касаться внутренней поверхности фильтра. Фильтр в воронке может быть заполнен жидкостью только так, чтобы ее уровень находился на 0,5 см ниже края фильтра. Стеклянную палочку после каждого приливания жидкости опускают в стакан.
3. Промывание осадка на фильтре проводят небольшими порциями промывной жидкости из промывалки, смывая струей жидкости осадок со стенок фильтра в его нижнюю часть. Общий объем промывной жидкости не должен превышать 100 мл. При добавлении промывной жидкости небольшими порциями к осадку достигается более полное удаление адсорбированных примесей с поверхности осадка. 4. При переведении осадка в гравиметрическую форму могут протекать побочные реакции. При озолении фильтра: ВаSO4 + 2С = 2СО2 + ВаS Обратный переход сульфида бария в сульфат происходит при длительном нагревании осадка на воздухе: BaS + 2O2 = BaSO4 Возможно также последующее превращение BaS в ВаСО3 под действием СО2 и влаги воздуха. При слишком высокой температуре прокаливания (>800°С): BaSO4 = BaO + SO3
Контрольные вопросы
1. Сущность гравиметрического анализа: растворимость осадков и ее влияние на анализ, свойства осадков, разделение фаз. 2. Расчет растворимости (моль/л, г/л) исходя из величины ПР. 3. Осаждаемая и гравиметрическая формы; требования, предъявляемые к осаждаемой форме. 4. Факторы, влияющие на полноту осаждения и выбор осаждающего реагента (произведение растворимости и произведение активностей, константа равновесия гетерогенной реакции). 5. Теоретическое обоснование выбора оптимальных условий осаждения кристаллических и аморфных осадков, механизм образования осадков. 6. Виды загрязнений осадков и способы их очистки, фильтрование и промывка осадков. 7. Получение гравиметрической формы и требования, предъявляемые к ней. 8. Техника гравиметрического анализа, конкретные примеры определений. 9. Вычисления в гравиметрии. Аналитические возможности метода. ТИТРИМЕТРИЯ Титрование – метод анализа, предложенный Ж.Л.Гей-Люссаком в XIX веке. Он получил широкое распространение за точность скорость и простоту. Титриметрический метод основан на определении количества реагента (титранта), затраченного на реакцию с определяемым веществом. При выполнении анализа к точно измеренному объему анализируемого раствора постепенно прибавляют непрерывно контролируемый объем реагента с точно известной концентрацией до того момента, когда количество эквивалентов титранта станет равным количеству эквивалентов определяемого вещества. Этот момент называется точкой эквивалентности. Уравнение реакции титрования обязательно должно быть известным А + В = C + D Тогда условие эквивалентности
или
Откуда рассчитывают неизвестную концентрацию определяемого вещества. Точку эквивалентности определяют с помощью специальных индикаторов, резко изменяющих окраску в этот момент. При достижении точки эквивалентности титрование останавливают и записывают объем раствора титранта. Для титриметрических определений можно использовать реакции различных типов: кислотно-основного взаимодействия, окислительно-восстановительные, комплексообразования, осаждения. Ко всем реакциям предъявляются общие требования: – строгое соответствие стехиометрическому уравнению; – количественное протекание, степень полноты не менее 99,9%; – высокая скорость взаимодействия. Для проведения титрования используют специальную посуду и приемы работы. Мерные колбы. Мерные колбы (рис. 1) – круглые плоскодонные колбы с узким длинным горлом, приблизительно на середине которого находится круговая метка. Если налить жидкость так, чтобы нижний мениск ее находился на уровне метки, тогда объем находящейся в колбе жидкости будет равен номинальной вместимости колбы. Мерные колбы калиброваны при 20 °С на вливание. На стенке колбы находится марка завода-изготовителя, в которой указаны номинальный объем колбы, температура калибрования, класс точности, ГОСТ. Мерные колбы изготовляют вместимостью 25, 50, 100, 200, 250, 500, 1000, 2000 см3. Они имеют либо пришлифованные стеклянные пробки, либо закрываются резиновыми пробками. Мерные колбы предназначены для приготовления растворов точно известного объема либо путем разбавления точно измеренного объема (аликвотной части) раствора более высокой концентрации, либо растворением точной навески вещества в дистиллированной воде и других растворителях. Объем раствора в мерной колбе доводят до метки следующим образом: из промывалки наливают растворитель до уровня приблизительно на 1 см ниже метки, затем при помощи пипетки с резиновым колпачком по каплям добавляют растворитель до метки. При этом глаза наблюдателя должны находиться на уровне метки на колбе. Мерную колбу нужно всегда брать за горло выше уровня раствора, в противном случае колба и раствор за счет тепла руки нагреваются, и объем раствора не будет соответствовать номинальному. Пипетки. Пипетки (рис. 1, б) предназначены для точного отмеривания определенного объема жидкости и переноса ее из одного сосуда в другой (отбор аликвотных частей). Эту операцию производят следующим образом. Чистую пипетку берут большим и средним пальцами правой руки за ее верхний конец, погружают нижний конец на 3 – 4 см в жидкость и всасывают ее приблизительно до половины пипетки. Быстро закрывают верхнее отверстие пипетки указательным пальцем, вынимают пипетку из жидкости, наклоняют пипетку почти горизонтально и, не отпуская указательный палец, поворачивают ее так, чтобы жидкость обмыла стенки, затем выливают ее. Ополаскивание пипетки повторяют. Для отбора аликвотной части погружают конец пипетки в жидкость, всасывают ее до уровня немного выше метки и закрывают верхнее отверстие указательным пальцем. Затем приподнимают пипетку над жидкостью и, ослабив нажим указательного пальца, медленно, по каплям, выпускают избыток жидкости до тех пор, пока нижний край мениска не коснется метки (глаза должны находиться на одном уровне с меткой), в этот момент плотно прижимают указательный палец и прекращают тем самым вытекание жидкости. Если на кончике пипетки остается капля, ее удаляют прикосновением конца пипетки к стенке сосуда. Для выливания жидкости из пипетки опускают в соответствующий сосуд нижний конец пипетки, открывают верхнее отверстие и дают жидкости свободно вытечь, затем слегка касаются носиком пипетки дна или стенки сосуда, держат так пипетку приблизительно 10 с и вынимают ее из сосуда. В носике пипетки всегда остается жидкость. Операция отбора аликвотных частей жидкости должна быть отработана до уровня навыка, это гарантирует ее правильность и воспроизводимость. Заполнять пипетку ядовитыми жидкостями, концентрированными кислотами и щелочами надо только с помощью специальных приспособлений (груш и др.). Бюретки. Бюретки (рис. 3) – длинные стеклянные трубки с краном или другим запорным устройством. Это может быть надетая на конец бюретки резиновая трубка со стеклянным оттянутым наконечником, которая либо пережимается металлическим зажимом, либо в нее вставлен стеклянный шарик, служащий затвором. При сдавливании резиновой трубки в месте расположения шарика сверху вниз по касательной резина оттягивается, образуется щель между шариком и внутренней стенкой резиновой трубки, через которую вытекает раствор из бюретки. На наружной стенке бюретки нанесена шкала из больших делений, соответствующих целым миллилитрам, и малых, соответствующих десятым долям миллилитра.
Рис. 2. Бюретки
Бюретки предназначены для измерения объема вылитой из них жидкости, поэтому они калиброваны на выливание. Бюретки используют главным образом при титровании. Подготовка бюретки к титрованию. Чистую бюретку, закрепленную в лапке лабораторного штатива, ополаскивают 2 – 3 раза раствором титранта, для чего через воронку наливают его каждый раз по 5 – 7 см3 в бюретку и выливают через запорное устройство в коническую колбу или стакан. Затем наливают в бюретку через воронку раствор титранта до уровня на 2 – 3 см выше отметки «0» и заполняют им носик бюретки. Для этого перегибают резиновую трубку, соединяющую бюретку с носиком так, чтобы его конец был направлен вверх, и надавливают кончиками большого и указательного пальцев на стеклянный шарик, находящийся внутри трубки. При этом раствор титранта, вытесняя воздух, заполняет носик бюретки; вытекающий из носика раствор собирают в колбу или стакан для слива. После этого вынимают из бюретки воронку (не забывать!) и выпускают через затвор по каплям титрант в посуду для слива до тех пор, пока нижний край мениска раствора не коснется нулевой отметки бюретки (глаза должны быть на уровне отметки). Титрование. Коническую колбу с аликвотной частью титруемого раствора подставляют под носик бюретки и приподнимают за горло так, чтобы конец носика находился в конической части колбы. Кончиками большого и указательного пальцев другой руки, надавливая на запорный стеклянный шарик, приливают раствор титранта в колбу, регулируя скорость приливания силой давления пальцев на шарик. Прекращают приливать раствор титранта в момент, указанный в методике. Во время титрования раствор в колбе непрерывно перемешивают легким круговым движением колбы. Расход титранта отсчитывают по нижнему краю мениска титранта в бюретке, определяя на глаз сотые доли миллилитра. Результат титрования записывают в рабочий журнал, обязательно указывая сотые доли см3, например 15,46 см3; 9,70 см3 и т.п. Параллельно титруют 4 – 5 проб и находят средний расход титранта, при этом разность любых двух титрований не должна превышать 0,05 см3. Перед каждым титрованием, необходимо заполнять бюретку раствором титранта до нулевой отметки. Если титрант интенсивно окрашен, его уровень определяют по верхнему краю мениска. Примечание Нет необходимости добиваться того, чтобы масса навески совпадала с расчетной величиной g, как и не следует стремиться к тому, чтобы тетраборат натрия до последней крупинки высыпался из бюкса в мерную колбу. Необходимо не допустить потери даже самого малого количества навески и важно точно знать ее массу. По взятой навеске тетрабората натрия и вместимости мерной колбы рассчитать С и Т раствора тетрабората натрия. Цель работы Определить массу NaOH и Na2CO3, находящихся в растворе методом кислотно-основного титрования при совместном присутствии (последовательное титрование). Определение компонентов смесей NaOH + Na2CO3 в растворе основано на титровании его стандартным раствором НС1 сначала в присутствии индикатора фенолфталеина и затем – метилового оранжевого. При титровании раствора смеси NaОН + Na2СО3 по фенолфталеину фиксируется момент титрования, когда оттитрованы NaOH и Na2СО3 до NaНСО3, расход титраита при этом составит V 1. При титровании этого раствора по метиловому оранжевому NaНСО3 титруется до угольной кислоты, общий расход титранта составит V2, причем 2V1 > V2. Объем титранта (V2 – V1) эквивалентен содержанию Na2СО3, f экв(Na2СО3) = 1, a V2 – 2(V2 – V1) эквивалентен содержанию NaOH. Для пояснения сказанного титрование раствора смеси NaOH + Na2CO3 схематично изображено на рис. 2
Рис.2. Схема титрования смеси Na2CO3 и NaOH
Реактивы и оборудование. Коническая колба для титрования объемом 250 см3; пипетка на 10,00 см3; бюретка объемом 25 см3; мерная колба на 100,00 см3. Титрант – стандартный раствор HCl c концентрацией C(HCl) = 0,10 н; кислотно-основной индикатор метиловый оранжевый 1% раствор, кислотно-основной индикатор фенолфталеин 1% раствор.
Выполнение работы. 1. Отбирают пипеткой аликвотные части раствора Vа из мерной колбы V, и переносят их в колбы для титрования, добавляют 3 капли раствора фенолфталеина и медленно при перемешивании титруют стандартным раствором НС1 до обесцвечивания розовой окраски; записывают расход титранта – V1. 2. Затем добавляют в титруемый раствор 1 – 2 капли раствора метилового оранжевого и продолжают титрование до перехода окраски раствора из желтой в оранжевую, записывают расход титранта – V2.
Задание
1. По результатам титрований находят средние значения m(Na2CO3) и m(NaOH) из серии параллельных определений.
2. Рассчитать абсолютную и относительную ошибку определения, сравнив полученные результаты, с истинным значением, выданным лаборантом. Абсолютная ошибка составляет: d = m – mист. Относительную ошибку определения вычисляют по формуле
Контрольные вопросы 1. Дайте определение понятию «массовая доля растворенного вещества»; молярная концентрация; молярная концентрация эквивалента (эквивалентная концентрация). 2. Вычислите молярные массы эквивалентов и факторы эквивалентности в реакциях полной нейтрализации следующих веществ: HNO3; NH3; H2SO4; KHSO4; Na2CO3. 3. Рассчитайте объем 0,1 н. HNO3, необходимой для нейтрализации 5,3 г Na2CO3. 4. Расчет рН слабых и сильных кислот и оснований в водных растворах. 5. Буферные растворы, используемые в химическом анализе, их состав, свойства, расчет рН. Буферная емкость. 6. Использование реакций гидролиза в анализе. Принцип расчета рН гидролизующихся солей. 7. Кривые кислотно-основного титрования, расчет и построение теоретических кривых титрования сильных и слабых кислот и оснований. 8. Скачок кислотно-основного титрования. Способы индикации конечной точки титрования. 9. Кислотно-основные индикаторы, механизм изменения их окраски. Интервал перехода окраски индикатора. Показатель титрования рТ. Правило выбора индикатора по теоретическим кривым титрования. Примеры двухцветных и одноцветных индикаторов. Опыт 1. Приготовление ЭДТА В качестве титранта в методе комплексонометрического титрования применяют стандартный раствор ЭДТА. Раствор готовят из двунатриевой соли ЭДТУ (ЭДТА, трилон Б, комплексон III), кристаллизующейся с двумя молекулами воды. Ее состав отвечает формуле Na2C10H14N2×2H2O. Молекулярная масса этого соединения составляет 372,242 у. е. Применение натриевой соли этилендиаминтетрауксусной кислоты для приготовления стандартного раствора обусловлено ее лучшей растворимостью в воде по сравнению с кислотой. Так, при 22 оС растворимость ЭДТА составляет 10,8 г на 100 г воды, а ЭДТУ только 0,2 г на 100 г воды. Для выполнения лабораторных работ готовят 500 мл 0,025 М раствора ЭДТА. Навеска, необходимая для приготовления указанного объема стандартного раствора ЭДТА, составляет:
Выполнение работы. 1. Навеску ЭДТА 4 – 5 г взвешивают в бюксе на технических весах, переносят в колбу вместимостью 500 мл и доводят до метки дистиллированной водой. 2. Бюкс после перенесения навески взвешивают на технических весах и по разности двух взвешиваний находят массу навески ЭДТА. Раствор в колбе тщательно перемешивают, растворение ЭДТА происходит медленно.
ПРИЛОЖЕНИЯ Литература 1. Васильев, В.П. Аналитическая химия: учеб. для вузов по хим.-технол. спец. Кн. 1 - М.: Дрофа, 2009. - 366, [2] с. - (84623-12) (543; В 19) и предыдущие издания. 2. Васильев, В.П. Аналитическая химия [Текст]: учеб. для вузов по хим.-технол. спец. Кн. 2 - М.: Дрофа, 2009. - 383 с. - (74072-35) (543; В 19) (84625-12) и предыдущие издания. 3. Алексеев, В.Н. Количественный анализ: учебник для нехим. спец. вузов - М.: Химия, 1973. - 594 с. - (85023-1) (544; А 47) 4. Крешков, А.П. Основы аналитической химии: учебник для хим.-технол. спец. вузов. Кн. 1 - М.: Химия, 1976. - 472 с. - (51933-1) (543; К 80)
ГРАВИМЕТРИЯ
Лабораторная работа № 1
Цель работы. Определение воды в твердых веществах – один из наиболее важных случаев определения, так как вода нередко является основной частью анализируемых веществ. Содержание воды определяют как прямыми, так и косвенными методами. Кристаллизационной называется вода, входящая в структуру кристаллов некоторых веществ, называемых кристаллогидратами. Содержание кристаллизационной воды в кристаллогидратах отвечает определенным химическим формулам, например ВаС12×2Н2О, CuSO4×5H2O, Na2SO4×10H2O, Na2SO4×7H2O и т. д. Вследствие этого кристаллизационную воду называют также стехиометрической водой. При нагревании кристаллогидраты разлагаются с выделением воды. На этом основано определение содержания кристаллизационной воды в большинстве кристаллогидратов методом отгонки. Так, в рассматриваемом случае навеску ВаС12×2Н2О, помещенную в бюкс, нагревают до 120 – 125 °С в сушильном шкафу до тех пор, пока не перестанет изменяться масса вещества (высушивание до постоянной массы). Достижение постоянной массы свидетельствует, очевидно, о том, что вся кристаллизационная вода уже удалена. Масса ее равна убыли в массе вещества.
Приборы и реактивы. Сушильный шкаф; технические весы; аналитические весы; эксикатор; тигельные щипцы; стеклянный бюкс; хлорид бария кристаллический, BaCl2×2H2O.
Выполнение работы. Взятие навески. 1. Тщательно вымытый бюкс высушивают в сушильном шкафу и, не закрывая крышкой, охлаждают, поставив на 20 мин в эксикатор около весов. 2. После этого, соблюдая все правила, точно взвешивают бюкс вместе с крышкой на аналитических весах. Затем помещают в него около 1,5 г свежеперекристаллизованного ВаС12×2Н2О и, закрыв крышкой, снова точно взвешивают. Высушивание. 1. Открыв бюкс, помещают крышку сверху бюкса боком (т. е. перевернув на ребро) и ставят на полку (не на дно) сушильного шкафа. Под бюкс следует подложить листок бумаги с указанием своей фамилии. Выдерживают бюкс в шкафу при температуре около 125 °С приблизительно 2 ч. Затем тигельными щипцами переносят бюкс и крышку его в эксикатор. 2. Продержав эксикатор 20 мин около весов, вынимают из него бюкс и, закрыв крышкой, точно взвешивают. Далее снова ставят бюкс с веществом в сушильный шкаф и выдерживают его там (открыв крышку) еще около 1 ч, повторно охлаждают в эксикаторе и снова взвешивают. 3. Если второе взвешивание дает тот же самый результат, что и первое, или отличается от него не более чем на 0,0002 г, кристаллизационную воду можно считать удаленной практически полностью. В противном случае высушивание с периодическим взвешиванием повторяют до тех пор, пока не будет достигнуто постоянство массы. Результаты всех повторных взвешиваний обязательно записывают в лабораторный журнал, даже если они одинаковые. Если работу приходится прерывать, бюкс оставляют в эксикаторе.
Таблица. Результаты определения кристаллизационной воды
Расчеты. Масса кристаллизационной воды в навеске: m(H2O) = 9,5895 - 9,3748 = 0,2147 г Массовая доля кристаллизационной воды в ВаС12×2Н2О вычисляют из пропорции: в 1,4575 г ВаС12×2Н2О содержится 0,2147 г Н2О в 100 г ВаС12×2Н2О – х г Н2О
Расчет ошибки определения. Для проверки найденную величину сравнивают с теоретически вычисленным значением массовой доли Н2О в ВаС12×2Н2О. В 1 моль (244,3 г) ВаС12×2Н2О содержится 2 моль воды, т.е. 36,03 г Н2О. Составим пропорцию: в 244,3 г ВаС12×2Н2О содержится 36,03 г Н2О в 100 г ВаС12×2Н2О – х г Н2О
Абсолютная ошибка составляет: d = 14,73% – 14,75% = –0,02%. Относительную ошибку определения вычисляют по формуле
Эта погрешность объясняется ошибками взвешивания. При правильной работе абсолютная ошибка данного определения не должна превышать ±0,05%. Другими словами, приемлемы результаты, лежащие в пределах от 14,70 до 14,80%.
Лабораторная работа № 2
Метод основан на осаждении сульфат-ионов в виде кристаллического белого осадка BaSO4 и получении гравиметрической формы (BaSO4) прокаливанием: SO42– + Ba2+ = BaSO4 Данным методом определяют сульфатную серу, а также свободную, пиритную и сульфидную, предварительно окислив их до сульфатной.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
Последнее изменение этой страницы: 2016-12-10; просмотров: 1819; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.139.97.157 (0.165 с.) |
 %
% %
% %
%







 %
%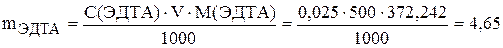 г
г


