
Заглавная страница Избранные статьи Случайная статья Познавательные статьи Новые добавления Обратная связь КАТЕГОРИИ: ТОП 10 на сайте Приготовление дезинфицирующих растворов различной концентрацииТехника нижней прямой подачи мяча. Франко-прусская война (причины и последствия) Организация работы процедурного кабинета Смысловое и механическое запоминание, их место и роль в усвоении знаний Коммуникативные барьеры и пути их преодоления Обработка изделий медицинского назначения многократного применения Образцы текста публицистического стиля Четыре типа изменения баланса Задачи с ответами для Всероссийской олимпиады по праву 
Мы поможем в написании ваших работ! ЗНАЕТЕ ЛИ ВЫ?
Влияние общества на человека
Приготовление дезинфицирующих растворов различной концентрации Практические работы по географии для 6 класса Организация работы процедурного кабинета Изменения в неживой природе осенью Уборка процедурного кабинета Сольфеджио. Все правила по сольфеджио Балочные системы. Определение реакций опор и моментов защемления |
Химическая модификация белков. Задачи, решаемые c помощью химической модификации. Бифункциональные реагенты. Пост-трансляционная модификация белков в клетке.Стр 1 из 3Следующая ⇒
Химический синтез пептидов. Методы защиты функциональных групп. Методы создания пептидной связи. Представление о блочном и ступенчатом синтезе пептидов. Проблема рацемизации. Твердофазный синтез пептидов. Пептидный синтез — это построение пептидной цепи путем соединения аминокислот с помощью химических методов. Обычно речь идет о получении пептидов, содержащих до 40 — 45 аминокислот, таким способом можно осуществить синтез и небольших белков. Современный пептидный синтез располагает богатым арсеналом методических возможностей и представляет собой высокоразвитую область биоорганической химии. В общем случае синтез любого пептида состоит из трех основных стадий: блокирования (защиты) не участвующих в реакции функциональных групп аминокислоты или пептида; конденсации активированной карбоксильной группы одного компонента с аминогруппой другого; селективного или полного удаления защитных групп для продолжения синтеза или получения свободного пептида. Пептидный синтез служит надежным средством доказательства строения природных пептидно- белковых веществ. Синтетические пептиды широко используются для структурно-функциональных исследований. С помощью химических методов удается получать аналоги биологически активных пептидов, в том числе циклические производные с заданными свойствами (например, с пролонгированным, усиленным или избирательным действием), а также аналоги с остатками небелковых аминокислот. Синтетические пептидные фрагменты белков применяются для изучения их антигенных свойств и получения специфичных к отдельным участкам полипептидных цепей антител, используемых в структурно- функциональном анализе и в создании диагностикумов и вакцин. Методами пептидного синтеза получаются (в том числе и в промышленном масштабе) многие практически важные препараты для медицины и сельского хозяйства. В пептидном синтезе существуют два типа защитных групп — постоянные и временные. Постоянными называют группировки, используемые для защиты боковых функциональных групп и удаляемые на заключительном этапе синтеза пептида. Временными являются защитные группы для Nα - концевой аминогруппы и С- концевого карбоксила, снимаемые соответственно перед каждой стадией удлинения цепи или конденсации фрагментов. Защитные группы, используемые в синтезе пептидов, должны
удовлетворять следующим условиям: — полностью блокировать соответствующую группировку от участия в проводимых химических реакциях; — быть устойчивыми в ходе удаления других защитных групп; — не вызывать побочных реакций и рацемизации при введении, удалении и при образовании пептидных связей; — защищенные производные должны быть устойчивыми идентифицируемыми соединениями; — не вызывать осложнений с растворимостью и выделением пептидов из реакционных смесей. (примеры защитных групп см. в учебнике биоорганической химии) Образование пептидной связи в общем сводится к отщеплению элементов воды. Для того чтобы сделать эту реакцию возможной и, более того,
Хлорангидридный метод. Азидный метод. Метод ангидридов. Метод активированных эфиров. Карбодиимидный метод. Карбоксиангидридный метод.
Рацемизация, т. е. полная или частичная потеря оптической чистоты одного или более аминокислотных остатков, является главной побочной реакцией в пептидном синтезе, накладывающей жесткие ограничения на выбор защитных групп и методов конденсации. Рацемизация приводит к образованию оптически неоднородных продуктов, разделение которых по мере удлинения цепи резко осложняется и превращается в практически неосуществимую задачу. Реакция рацемизации протекает по двум механизмам: 1) через образование 5(4Н)- оксазолонов (часто называемых азлактонами) или 2) через енолизацию. В общем случае степень инверсии у α-углеродного атома (рацемизации) определяется природой заместителей X, R, Y, температурой и рН среды. Аминокислоты и их неактивированные производные заметно рацемизуются в сильнокислой или щелочной среде, особенно при нагревании. Активированные производные аминокислот более подвержены рацемизации как в процессе их получения, так и в ходе аминолиза. Особенно легко рацемизуются производные пептидов, что осложняет проведение конденсации фрагментов. Следует отметить, что уретановые N- защитные группировки аминокислот (в том числе наиболее популярные Z- и Вос- группы) обладают низкой склонностью к образованию оксазолонов. Поэтому ступенчатый
синтез с использованием этих групп — один из наиболее надежных путей избежать рацемизации при синтезе пептидов.
Синтез на полимерном носителе. Пептидный синтез в классическом варианте сопряжен со значительными затратами труда и времени. С целью создания более эффективной методологии Р. Меррифилд в 1963 г. предложил твердофазный метод синтеза пептидов. Идея его состоит в закреплении растущей полипептидной цепи на полимерном нерастворимом носителе. При этом значительно упрощаются операции выделения промежуточных продуктов, которые сводятся к экстракции и фильтрованию полимера, полностью снимается проблема нерастворимости пептидов и создаются предпосылки для автоматизации процесса. Определяющим фактором в твердофазном синтезе является полнота протекания всех химических реакций, которая достигается за счет применения избытка конденсирующего агента и N- защищенной аминокислоты, отделяемых экстракцией. Естественно, выбор защитных группировок и методов конденсации должен обеспечить полное отсутствие рацемизации. Наилучшие результаты достигаются при использовании Воc-, Врос- и Fmoc- защитных групп, методов симметричных ангидридов и дициклогексилкарбодиимидного. Успешное проведение синтеза на твердом полимере требует применения высокоочищенных реагентов и растворителей на всех стадиях процесса. Первый твердофазный синтез гормона брадикинина был проведен Р. Меррифилдом с общим выходом 70%. В качестве носителя наиболее широко используется микропористый хлорметилированный сополимер стирола и дивинилбензола, хорошо набухающий в органических растворителях и обладающий химической и механической прочностью.
3. Общая стратегия определения структуры белков. Анализ аминокислотного состава. Определение N- и С-концевых аминокислотных остатков. Фрагментация полипептидной цепи. Ферментативные и химические методы расщепления полипептидной цепи. Последовательная деградация белков по методу Эдмана. Определение аминокислотной последовательности белка с помощью автоматического секвенатора. Принципиально первичную структуру белков можно определять путем непосредственного анализа аминокислотной последовательности или путем расшифровки нуклеотидной последовательности соответствующих генов с помощью генетического кода. Естественно, наибольшую надежность обеспечивает сочетание этих методов. Исследование первичной структуры белка начинается с определения его молекулярной массы, аминокислотного состава, N- и С- концевых аминокислотных остатков. Поскольку пока не существует метода, позволяющего установить полную первичную структуру белка на целой молекуле, полипептидную цепь подвергают специфичному расщеплению химическими реагентами или протеолитическими ферментами. Смесь образовавшихся пептидных фрагментов разделяют и для каждого из них определяют аминокислотный состав и аминокислотную последовательность. После того как структура всех фрагментов установлена, необходимо выяснить порядок их расположения в исходной полипептидной цепи. Для этого белок подвергают расщеплению при помощи другого агента и получают второй, отличный от первого набор пептидных фрагментов, которые разделяют и анализируют аналогичным образом. Предположим, что исследуемый белок имеет последовательность, представленную на схеме. При действии на него трипсином
Анализ аминокислотного состава включает полный кислотный гидролиз исследуемого белка или пептида с помощью 5,7 н. соляной кислоты и количественное определение всех аминокислот в гидролизате. Гидролиз образца проводится в запаянных ампулах в вакууме при 110 °С в течение 24 ч. При этом полностью разрушается триптофан и частично серии, треонин, цистин и цистеин, а глутамин и аспарагин превращаются соответственно в глутаминовую и аспарагиновую кислоты. В то же время пептидные связи, образованные аминокислотными остатками с разветвленной боковой цепью (Val, Не, Leu), из- за стерических препятствий гидролизуются частично. Особенно стабильны связи Val—Val, He—Не, Val—Не и Не—Val. Для определения аминокислот используется нингидрин. Для повышения чувствительности вместо нингидрина в ряде анализаторов применяют флуорескамин или о- фталевый альдегид.
В полипептидной цепи белка с одной стороны расположен аминокислотный остаток, несущий свободную а- аминогруппу (амино- или N- концевой остаток), а с другой — остаток со свободной а- карбоксильной группой (карбоксильный, или С- концевой остаток). Анализ концевых остатков играет важную роль в процессе определения аминокислотной последовательности белка. На первом этапе исследования он дает возможность оценить число полипептидных цепей, составляющих молекулу белка, и степень гомогенности исследуемого препарата. На последующих этапах с помощью анализа N- концевых аминокислотных остатков осуществляется контроль за процессом разделения пептидных фрагментов.
остатков лизина и тирозина могут получаться е- ДНС- лизин и О- ДНС- тирозин. ДНС- Аминокислоты обладают интенсивной флуоресценцией в ультрафиолетовой области спектра (Х ВО3 6. = 365 нм); обычно для их идентификации достаточно 0,1 — 0,5 нмоль вещества. Имеется ряд методов, с помощью которых можно определять как N- концевой аминокислотный остаток, так и N- концевую аминокислотную последовательность. К ним относятся деградация по методу Эдмана и ферментативный гидролиз аминопептидазами.
аминокислоты, что затрудняет интерпретацию результатов. Оксазолоновый метод, часто называемый методом тритиевой метки, основан на способности С- концевого аминокислотного остатка под действием уксусного ангидрида подвергаться циклизации с образованием оксазолона. В щелочных условиях резко увеличивается подвижность атомов водорода в положении 4 оксазолонового кольца, и он может быть легко обменен на тритий. Образующиеся в результате последующего кислотного гидролиза тритиированного пептида или белка продукты реакции содержат радиоактивно меченную С- концевую аминокислоту. Хроматографирование гидролизата и измерение радиоактивности позволяют идентифицировать С- концевую аминокислоту пептида или белка.
Необходимый этап в определении первичной структуры белка — расщепление белковой молекулы на пептидные фрагменты. Химические методы (бромциан, щепит по остаткам метионина, N-бромсукцинимид щепит по остаткам триптофана) обладают высокой селективностью, однако процесс расщепления протекает, как правило, с выходом, не превышающим 50%. Эти методы целесообразно использовать для получения крупных фрагментов. Возможности ферментативных методов гидролиза значительно шире, они дают как крупные, так и мелкие пептиды.
Ферментативные методы гидролиза. Наиболее широко используемым ферментом при установлении первичной структуры белков является трипсин. Фермент обладает уникальной субстратной специфичностью, катализируя гидролиз связей, образованных карбоксильными группами только основных аминокислот — лизина и аргинина. Однако в ряде случаев на полноту и скорость протекания процесса оказывают влияние положение гидролизуемой связи в цепи и химическая природа боковых групп соседних аминокислотных остатков. Наряду с трипсином из поджелудочной железы выделяют другую сериновую протеиназу — химотрипсин. Химотрипсин обладает гораздо более широкой специфичностью, чем трипсин. Фермент преимущественно катализирует гидролиз пептидных связей, образованных карбоксильными группами ароматических аминокислот — тирозина, фенилаланина и триптофана. С меньшей скоростью гидролизуются пептидные связи лейцина, метионина, гистидина. Скорость расщепления отдельных связей в белках и пептидах зависит от характера соседних аминокислотных остатков. В последнее время при исследовании первичной структуры белков широкое применение находит протеиназа из Staphylococcus aureus, выделенная в 1972 г. Г. Р. Драпю, которая также относится к классу сериновых протеиназ. Протеиназа из S. aureus с высоким выходом расщепляет пептидные связи, образованные карбоксильной группой глутаминовой кислоты. Термолизин, выделяемый из культуральной среды термофильной бактерии Bacillus thermoproteolyticus, относится к классу нейтральных протеиназ, содержащих цинк в качестве кофактора. Термолизин необычайно термостабилен: в течение часа он полностью сохраняет свою активность при 60 °С (рН 7,0) и теряет менее 50% активности при 80 °С. В отличие от большинства протеолитических ферментов, специфичность термолизина определяется природой остатка, которому принадлежит аминогруппа гидролизуемой связи. Термолизин преимущественно расщепляет пептидные связи, включающие аминокислотные остатки с гидрофобной боковой цепью (Ilе, Leu, Val, Phe, Туг, Trp).
Для установления аминокислотной последовательности белков используется совокупность химических, ферментативных и физико-химических методов. Метод Эдмана. Основным методом определения аминокислотной последовательности является метод деградации полипептидной цепи с помощью фенилизотиоцианата (ФИТЦ), разработанный П. Эдманом в 1950 — 1956 гг. Метод Эдмана позволяет последовательно отщеплять N- концевые аминокислотные остатки в виде фенил- тиогидантоинов (ФТГ). Каждый цикл деградации включает три стадии: 1) образование фенилтиокарбамоил (ФТК)- пептида, 2) отщепление N- концевого остатка аминокислоты в форме анилинотиазолинона, 3) изомеризацию тиазолинона в ФТГ и идентификацию последнего. Среди модификаций метода Эдмана широкое применение нашел метод, сочетающий последовательную деградацию пептида с анализом N- концевых аминокислотных остатков в виде их дансильных производных (ДНС- Эдман). По этому методу перед каждым циклом деградации отбирается определенная аликвотная часть пептида для анализа N- концевой аминокислоты (рис. 17). Достоинства метода — более высокая чувствительность определения ДНС-аминокислот и меньшие потери материала на каждой стадии деградации за счет исключения экстракции бензолом ФТК- производных пептидов.
Ферментативные методы определения аминокислотной последовательности. Для определения структуры пептидов и белков можно применять ферменты, катализирующие отщепление N- и С- концевых аминокислотных остатков полипептидной цепи (аминопептидазы и карбоксипептидазы). Гидролиз пептидов с помощью карбоксипептидаз является основным способом определения С- концевого остатка и С- концевой аминокислотной последовательности. При исследовании структуры пептидов и белков используются карбоксипептидазы А, В, С и Y. Карбоксипептидазы А (СРА) и В (СРВ) выделяют из поджелудочной железы крупного рогатого скота, карбоксипептидазу С (СРС) — из кожуры и листьев цитрусовых, карбоксипептидазу Y (CPY) —из пекарских дрожжей. Общим требованием к субстрату для всех карбоксипептидаз является наличие а- карбоксильной группы у С- концевой амино- кислоты. Природа боковой цепи у отщепляемого аминокислотного остатка — основной фактор, определяющий скорость гидролиза пептидной связи. Пептидгидродазы, отщепляющие N- концевые аминокислотные остатки пептидов и белков, составляют группу аминопептидаз. Необходимым условием для действия аминопептидаз является наличие у субстрата свободной а- аминогруппы. Ферменты этой группы гидролизуют и дипептиды. В структурных исследованиях белков наибольшее применение нашли лейцинаминопептидаза (ЛАП) и аминопептидаза М (АПМ), выделяемые из почек свиньи.
Масс- спектрометрический метод. Наряду с химическими и ферментативными методами для определения аминокислотной последовательности пептидов находят применение физико- химические методы, в частности масс- спектрометрия. Процесс съемки масс- спектра соединения состоит из нескольких стадий: переведение исследуемого образца в газообразное состояние; ионизация его, при которой происходит распад большинства образующихся молекулярных ионов; ускорение полученных ионов в электрическом поле, последующее их разделение (в зависимости от отношения массы к заряду) в магнитном поле; и наконец регистрация масс- спектра.
Крупным достижением в области структурных исследований белков явилось создание в 1967 г. П. Эдманом и Дж. Бэггом секвенатора — прибора, который с высокой эффективностью осуществляет последовательное автоматическое отщепление N- концевых аминокислотных остатков по методу Эдмана. В секвенаторе все реакции проводятся в цилиндрическом стеклянном стаканчике, вращающемся с постоянной скоростью в атмосфере инертного газа. Исследуемый образец белка наносится в виде тонкой пленки на стенки стаканчика, а реактивы и растворители по шлангу подаются на его дно. За счет центробежной силы они поднимаются по стенкам, соприкасаются с пленкой белка, затем собираются в специальной канавке в верхней части стаканчика и удаляются по отводному шлангу. Большая поверхность соприкосновения между двумя фазами способствует легкому проникновению реагентов сквозь пленку белка, быстрому протеканию реакций и беспрепятственной экстракции продуктов реакции. В секвенаторе исключен контакт анализируемого образца белка с кислородом воздуха и стандартизованы условия проведения всех стадий реакции. Наряду с тщательной очисткой используемых реагентов и растворителей, это позволяет проводить автоматическое
В 1981 г. Л. Худ и М. Ханкепиллер сконструировали новую модель секвенатора — газофазный секвенатор, предназначенный для анализа микроколичеств белков и пептидов. В приборе исследуемый образец наносится на небольшой (диаметр 10 мм) диск из пористого стеклянного волокна, пропитанный полибреном. После высушивания диск зажимается между двумя стеклянными цилинд- рами, образующими миниатюрную реакционную камеру. Через капилляр в центре верхнего цилиндра в реакционную камеру подаются необходимые реагенты (летучие — в газообразном состоянии), которые, проходя через поры диска, взаимодействуют с адсорбированным на нем белком или пептидом и далее выводятся через капилляр в нижнем цилиндре. Нековалентная иммобилизация образца на пористой пластинке, уменьшение объема реакционной камеры и соответственно расхода реагентов и растворителей для промывок позволили сократить до минимума потери материала в процессе деградации на газофазном секвенаторе и тем самым снизить количество анализируемого пептида или белка до уровня 100 — 500 пмоль. Четвертичная структура белков. Примеры гомомерных и гетеромерных белков. Методы исследования четвертичной структуры Многие белки состоят из субъединиц, одинаковых или различных, образующих трехмерные ассоциаты или более сложные ансамбли. В этом случае принято говорить о четвертичной структуре белков. Специфичность четвертичной структуры данного белка обусловливается выполняемой им биологической функцией, а взаимодействие субъединиц обеспечивает дополнительный механизм ее регуляции. Примеры гомомерных белков: глутаминсинтетаза (12 субъединиц образуют два гексагональных кольца), белок оболочки вируса табачной мозаики (2130 субъединиц)б микрофиламенты (актин). Примеры гетеромерных белков: гемоглобин (α2β2), аспартат-транскарбамоилаза (12 субъединиц — 6 каталитических (С- субъединицы) и 6 регуляторных (R- субъединицы), С- субъединицы объединены в тримеры, расположенные один над другим, а R-субъединицы находятся на периферии молекулы, взаимодействуют друг с другом и с С- субъединицами), ДНК-зависимая РНК-полимераза (2 α, 1 β, 1 β’, 1 δ субъединицы), нуклеосомы (гистоны H2A, H2B, H3, H4), микротрубочки (α и β субъединицы тубулина). Первый этап исследования четвертичной структуры белка — анализ его субъединичного состава. Для этого необходимо провести диссоциацию белка на отдельные субъединицы, что обычно достигается с помощью гидрохлорида гуанидина, мочевины или додецилсульфата натрия (с добавлением меркаптоэтанола для восстановления дисульфидных связей). Во многих случаях диссоциации способствуют изменение рН среды, добавление соли, замена воды на органические растворители, а также химическая модификация белка. Субстраты, различные метаболиты и кофакторы в разных белках влияют как на ассоциацию, так и диссоциацию субъединиц. Субъединичный состав белка может быть выяснен путем сопоставления его суммарной молекулярной массы с молекулярными массами отдельных субъединиц (установленными с помощью электрофореза в полиакриламидном геле в присутствии додецилсульфата натрия), а также с числом N- и С- концевых аминокислотных остатков. Другой метод анализа заключается в сравнении числа пептидов (N), ожидаемых на основании данных аминокислотного состава, с числом реально образующихся при том или ином специфическом гидролизе молекулы белка. Процесс формирования четвертичной структуры белков проходит в соответствии со строгими законами термодинамики. Силы, способствующие ассоциации белка, должны быть скомпенсированы таким образом, чтобы активные группировки, расположенные в областях, имеющих тенденции к ассоциации, не оставались экспонированными наружу. В белках, построенных из одинаковых субъединиц, последние практически всегда находятся в эквивалентном (или идентичном) окружении. Взаимодействие между комплементарными участками субъединиц в четвертичной структуре осуществляется с помощью ван-дер-ваальсовых сил, ионных и водородных связей. Формирование четвертичной структуры из трех и более субъединиц протекает с промежуточным образованием агрегатов меньшего размера. Агрегация первых мономеров часто облегчает присоединение последующих — в этом смысле образование четвертичной структуры является кооперативным процессом. Имеется ряд физико- химических методов исследования геометрии субъединиц в белках, обладающих четвертичной структурой. Наиболее информативный среди них — метод рентгеноструктурного анализа. Классическими являются рентгеноструктурные исследо- вания гемоглобина, проводившиеся в Кембридже М. Перутцем и сотр. в течение 25 лет, начиная с 1937 г. Была установлена субъединичная структура гемоглобина — α2β2. С каждой субъединицей связано по одной гем-группе, атом железа которой может обратимо связывать молекулу О 2. Контакты образуются преимущественно между α- и β- и в меньшей степени между α—α- и β—β- цепями белка. Большинство контактов возникает за счет взаимодействия гидрофобных боковых цепей аминокислотных остатков, часть из них обусловлена водородными и электростатическими связями. Электронная микроскопия — информативный метод анализа четвертичной структуры белков, особенно крупных молекул со многими субъединицами. Главные ограничения метода — сравнительная «прозрачность» белков по отношению к пучку электронов и их недостаточная стабильность в условиях эксперимента. Обычно удается проводить структурный анализ с разрешением до 2 нм, а при соблюдении определенных предосторожностей — до 1 нм. Совместное использование электронной микроскопии и рентгеноструктурного анализа дало возможность установить четвертичную структуру еще более сложно устроенного фермента — аспартат-транскарбамоилазы E.coli, катализирующего основную реакцию в биосинтезе пиримидиновых нуклеотидов — образование N- карба- моиласпарагиновой кислоты из аспарагиновой и карбамоилфосфор- ной кислот. Для исследования четвертичной структуры белков широко используется химическая модификация, в частности, бифункциональными реагентами. С помощью такого подхода была изучена пространственная структура ДНК- зависимой РНК- полимеразы E.coli. Проводилась модификация бифункциональными реагентами как самого фермента, так и его комплекса с фрагментами ДНК- матрицы, содержащими промоторные участки. В последнем случае использовались фрагменты ДНК, несущие фоточувствительные группировки (частично депуринизованные или содержащие остатки 5- бромурацила).
Возникает вопрос: почему многие белки состоят из субъединиц? Какие преимущества это дает по сравнению с одной длинной пептидной цепью? Во-первых, наличие субъединичной структуры позволяет «экономить» генетический материал. Для олигомерных белков, состоящих из идентичных субъединиц, резко уменьшается размер структурного гена и соответственно длина матричной РНК. Во- вторых, при сравнительно небольшой величине цепей уменьшается влияние случайных ошибок, которые могут возникнуть в процессе биосинтеза белковых молекул. В- третьих, наличие субъединичной структуры у многих белков позволяет клетке легко регулировать их активность, например, путем смещения равновесия ассоциация — диссоциация в ту или иную сторону. Наконец, субъединичная структура облегчает и ускоряет процесс молекулярной эволюции. Мутации, приводящие лишь к небольшим конформационным изменениям на уровне третичной структуры за счет многократного усиления этих изменений при переходе к четвертичной структуре, могут способствовать появлению у белка новых свойств. Характерной особенностью белков с четвертичной структурой является их способность к самосборке. Легко происходит, например, самосборка гемоглобина из смеси α- и β- цепей. Таким образом, в аминокислотной последовательности полипептидных цепей олиго- мерного белка закодированы как бы два уровня информации: один из них определяет трехмерную структуру отдельных полипептидных цепей, а второй, поскольку каждая субъединица содержит специфические участки связывания с другими субъединицами, определяет четвертичную структуру всей многокомпонентной молекулы в целом.
Химический синтез пептидов. Методы защиты функциональных групп. Методы создания пептидной связи. Представление о блочном и ступенчатом синтезе пептидов. Проблема рацемизации. Твердофазный синтез пептидов. Пептидный синтез — это построение пептидной цепи путем соединения аминокислот с помощью химических методов. Обычно речь идет о получении пептидов, содержащих до 40 — 45 аминокислот, таким способом можно осуществить синтез и небольших белков. Современный пептидный синтез располагает богатым арсеналом методических возможностей и представляет собой высокоразвитую область биоорганической химии. В общем случае синтез любого пептида состоит из трех основных стадий: блокирования (защиты) не участвующих в реакции функциональных групп аминокислоты или пептида; конденсации активированной карбоксильной группы одного компонента с аминогруппой другого; селективного или полного удаления защитных групп для продолжения синтеза или получения свободного пептида. Пептидный синтез служит надежным средством доказательства строения природных пептидно- белковых веществ. Синтетические пептиды широко используются для структурно-функциональных исследований. С помощью химических методов удается получать аналоги биологически активных пептидов, в том числе циклические производные с заданными свойствами (например, с пролонгированным, усиленным или избирательным действием), а также аналоги с остатками небелковых аминокислот. Синтетические пептидные фрагменты белков применяются для изучения их антигенных свойств и получения специфичных к отдельным участкам полипептидных цепей антител, используемых в структурно- функциональном анализе и в создании диагностикумов и вакцин. Методами пептидного синтеза получаются (в том числе и в промышленном масштабе) многие практически важные препараты для медицины и сельского хозяйства. В пептидном синтезе существуют два типа защитных групп — постоянные и временные. Постоянными называют группировки, используемые для защиты боковых функциональных групп и удаляемые на заключительном этапе синтеза пептида. Временными являются защитные группы для Nα - концевой аминогруппы и С- концевого карбоксила, снимаемые соответственно перед каждой стадией удлинения цепи или конденсации фрагментов. Защитные группы, используемые в синтезе пептидов, должны удовлетворять следующим условиям: — полностью блокировать соответствующую группировку от участия в проводимых химических реакциях; — быть устойчивыми в ходе удаления других защитных групп; — не вызывать побочных реакций и рацемизации при введении, удалении и при образовании пептидных связей; — защищенные производные должны быть устойчивыми идентифицируемыми соединениями; — не вызывать осложнений с растворимостью и выделением пептидов из реакционных смесей. (примеры защитных групп см. в учебнике биоорганической химии) Образование пептидной связи в общем сводится к отщеплению элементов воды. Для того чтобы сделать эту реакцию возможной и, более того,
Хлорангидридный метод. Азидный метод. Метод ангидридов. Метод активированных эфиров. Карбодиимидный метод. Карбоксиангидридный метод.
Рацемизация, т. е. полная или частичная потеря оптической чистоты одного или более аминокислотных остатков, является главной побочной реакцией в пептидном синтезе, накладывающей жесткие ограничения на выбор защитных групп и методов конденсации. Рацемизация приводит к образованию оптически неоднородных продуктов, разделение которых по мере удлинения цепи резко осложняется и превращается в практически неосуществимую задачу. Реакция рацемизации протекает по двум механизмам: 1) через образование 5(4Н)- оксазолонов (часто называемых азлактонами) или 2) через енолизацию. В общем случае степень инверсии у α-углеродного атома (рацемизации) определяется природой заместителей X, R, Y, температурой и рН среды. Аминокислоты и их неактивированные производные заметно рацемизуются в сильнокислой или щелочной среде, особенно при нагревании. Активированные производные аминокислот более подвержены рацемизации как в процессе их получения, так и в ходе аминолиза. Особенно легко рацемизуются производные пептидов, что осложняет проведение конденсации фрагментов. Следует отметить, что уретановые N- защитные группировки аминокислот (в том числе наиболее популярные Z- и Вос- группы) обладают низкой склонностью к образованию оксазолонов. Поэтому ступенчатый синтез с использованием этих групп — один из наиболее надежных путей избежать рацемизации при синтезе пептидов.
Синтез на полимерном носителе. Пептидный синтез в классическом варианте сопряжен со значительными затратами труда и времени. С целью создания более эффективной методологии Р. Меррифилд в 1963 г. предложил твердофазный метод синтеза пептидов. Идея его состоит в закреплении растущей полипептидной цепи на полимерном нерастворимом носителе. При этом значительно упрощаются операции выделения промежуточных продуктов, которые сводятся к экстракции и фильтрованию полимера, полностью снимается проблема нерастворимости пептидов и создаются предпосылки для автоматизации процесса. Определяющим фактором в твердофазном синтезе является полнота протекания всех химических реакций, которая достигается за счет применения избытка конденсирующего агента и N- защищенной аминокислоты, отделяемых экстракцией. Естественно, выбор защитных группировок и методов конденсации должен обеспечить полное отсутствие рацемизации. Наилучшие результаты достигаются при использовании Воc-, Врос- и Fmoc- защитных групп, методов симметричных ангидридов и дициклогексилкарбодиимидного. Успешное проведение синтеза на твердом полимере требует применения высокоочищенных реагентов и растворителей на всех стадиях процесса. Первый твердофазный синтез гормона брадикинина был проведен Р. Меррифилдом с общим выходом 70%. В качестве носителя наиболее широко используется микропористый хлорметилированный сополимер стирола и дивинилбензола, хорошо набухающий в органических растворителях и обладающий химической и механической прочностью.
|
|||||||||
|
|
Последнее изменение этой страницы: 2016-08-16; просмотров: 1203; Нарушение авторского права страницы; Мы поможем в написании вашей работы! infopedia.su Все материалы представленные на сайте исключительно с целью ознакомления читателями и не преследуют коммерческих целей или нарушение авторских прав. Обратная связь - 3.138.141.202 (0.093 с.) |
 обеспечить ее высокую скорость и полноту, необходимо «активировать» карбоксильную группу. Такая активация должна сводиться к увеличению электрофильности карбонильного углерода. Методы активации карбоксильных групп (более подробно см. учебник):
обеспечить ее высокую скорость и полноту, необходимо «активировать» карбоксильную группу. Такая активация должна сводиться к увеличению электрофильности карбонильного углерода. Методы активации карбоксильных групп (более подробно см. учебник):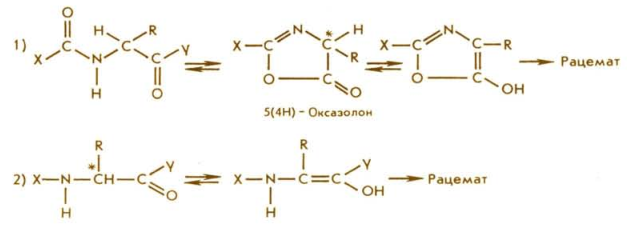

 гидролизуются связи Lys—Val и Arg—Ser, а при обработке бромцианом (BrCN) расщепляются связи Met—Туг и Met—Ala.
гидролизуются связи Lys—Val и Arg—Ser, а при обработке бромцианом (BrCN) расщепляются связи Met—Туг и Met—Ala. Один из первых методов определения N- концевых аминокислотных остатков был предложен Ф. Сенгером в 1945 г. При реакции а- аминогруппы пептида или белка с 2, 4- динитрофторбензолом получается динитрофенильное (ДНФ) производное, окрашенное в желтый цвет. Последующий кислотный гидролиз (5,7 н. НС1) приводит к разрыву пептидных связей и образованию ДНФ- производного N- концевой аминокислоты. ДНФ- Аминокислота экстрагируется эфиром и идентифицируется методом тонкослойной хроматографии в присутствии стандартов.
Один из первых методов определения N- концевых аминокислотных остатков был предложен Ф. Сенгером в 1945 г. При реакции а- аминогруппы пептида или белка с 2, 4- динитрофторбензолом получается динитрофенильное (ДНФ) производное, окрашенное в желтый цвет. Последующий кислотный гидролиз (5,7 н. НС1) приводит к разрыву пептидных связей и образованию ДНФ- производного N- концевой аминокислоты. ДНФ- Аминокислота экстрагируется эфиром и идентифицируется методом тонкослойной хроматографии в присутствии стандартов. Наибольшее применение для определения N- концевых остатков в настоящее время находит разработанный в 1963 г. В. Греем и Б. Хартли дансильный метод. Как и метод динитрофенилирования, он основан на введении в аминогруппы белка «метки», не удаляющейся при последующем гидролизе. Его первая стадия — реакция дансилхлорида (1-диметиламинонафталин- 5- сульфохлорида) с непротонированной а- аминогруппой пептида или белка с образованием дансилпептида (ДНС- пептида). На следующей стадии ДНС- пептид гидролизуется (5,7 н. НС1, 105 °С, 12 — 16 ч) и освобождается N- концевая а- ДНС- аминокислота. Кроме того, в результате дансилирования боковых групп
Наибольшее применение для определения N- концевых остатков в настоящее время находит разработанный в 1963 г. В. Греем и Б. Хартли дансильный метод. Как и метод динитрофенилирования, он основан на введении в аминогруппы белка «метки», не удаляющейся при последующем гидролизе. Его первая стадия — реакция дансилхлорида (1-диметиламинонафталин- 5- сульфохлорида) с непротонированной а- аминогруппой пептида или белка с образованием дансилпептида (ДНС- пептида). На следующей стадии ДНС- пептид гидролизуется (5,7 н. НС1, 105 °С, 12 — 16 ч) и освобождается N- концевая а- ДНС- аминокислота. Кроме того, в результате дансилирования боковых групп Среди химических методов определения С- концевых аминокислотных остатков заслуживают внимания метод гидразинолиза, предложенный С. Акабори, и оксазалоновый метод В. Матсуо. В первом из них при нагревании пептида или белка с безводным гидразином при 100 — 120 °С пептидные связи гидролизуются с образованием гидразидов аминокислот. С- Концевая аминокислота остается в виде свободной аминокислоты и может быть выделена из реакционной смеси и идентифицирована. Метод имеет ряд ограничений. При гидразинолизе разрушаются глутамин, аспарагин, цистеин и цистин; аргинин теряет гуанидиновую группировку с образованием орнитина. Гидразиды серина, треонина и глицина лабильны и легко превращаются в свободные
Среди химических методов определения С- концевых аминокислотных остатков заслуживают внимания метод гидразинолиза, предложенный С. Акабори, и оксазалоновый метод В. Матсуо. В первом из них при нагревании пептида или белка с безводным гидразином при 100 — 120 °С пептидные связи гидролизуются с образованием гидразидов аминокислот. С- Концевая аминокислота остается в виде свободной аминокислоты и может быть выделена из реакционной смеси и идентифицирована. Метод имеет ряд ограничений. При гидразинолизе разрушаются глутамин, аспарагин, цистеин и цистин; аргинин теряет гуанидиновую группировку с образованием орнитина. Гидразиды серина, треонина и глицина лабильны и легко превращаются в свободные

 отщепление аминокислотных остатков с выходом 95% и выше. В секвенаторе проводятся только первые две стадии реакции Эдмана — присоединение и отщепление. Образовавшиеся в результате анилинотиазолиноны экстрагируются и собираются в коллекторе фракций. Их превращение в ФТГ осуществляется вручную или с помощью автоматической приставки — конвертера.
отщепление аминокислотных остатков с выходом 95% и выше. В секвенаторе проводятся только первые две стадии реакции Эдмана — присоединение и отщепление. Образовавшиеся в результате анилинотиазолиноны экстрагируются и собираются в коллекторе фракций. Их превращение в ФТГ осуществляется вручную или с помощью автоматической приставки — конвертера.



